
促甲状腺素不敏感综合征(TSH insensitivity syndrome)又称为促甲状腺素(TSH)抵抗综合征(TSH resistance syndrome),是由于靶组织对TSH的敏感性下降,并导致甲状腺发育不良与甲状腺激素合成分泌降低而引起的先天性甲减。甲减的严重程度不均一,主要取决于TSH受体(TSHR)缺陷的严重性和功能代偿的程度。
TSH-β亚基/TSHR/Gsα缺陷导致TSH不敏感综合征
TSH不敏感遵循常染色体显性遗传模式,无不完全外显证据。尽管符合TSH不敏感表型的亚临床和轻度甲低在人群中常见,但遗传因素仍不明了,大部分家系无TSHR缺陷。目前,已经明确的TSH不敏感综合征病因有:①TSH-β亚基基因突变;②TSH受体基因突变;③Gsα亚基基因突变;④TSH受体信号转导途径缺陷(如转录因子PAX8基因突变)。
TSHR功能异常和甲状腺对TSH无反应是发病的中心环节
TSHR的配体结合区存在一定的特殊性,因为在此区至少存在下列4个特异性结合位点或结构域(下图):①TSH高亲和力结合部位可与TSH受体的拮抗剂或阻滞性TSH受体抗体结合;②TSH特异性结合部位与TSH高亲和力结合部位邻近;③LH/HCG结合部位的特异性不高,仅在血LH/HCG显著升高(如妊娠和胎盘滋养层细胞肿瘤)时起作用;④TSH低亲和力结合部位可与TSH受体激动剂或刺激性TSHR抗体结合。
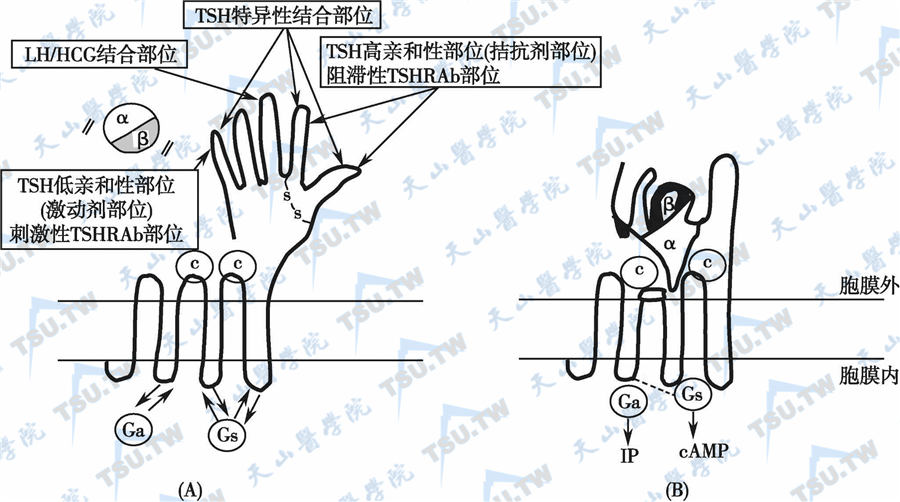
TSHR与配体结合前后的构象变化
注:(A):非激活状态下的TSHR的构象示意图,图中示TSHR的不同结合部位。(B):TSHR与TSH或其他配体(如TSHR抗体)结合后,将配体推向穿膜区的胞外环处,并激活G蛋白的信号转导通路,可能是TSH的β亚基与TSHR的胞外结合区(图中的“手”与“手指”)结合,而α亚基与TSHR的穿膜段互相作用,诱发TSHR空间结构变化
TSH受体缺陷
目前已发现两种缺陷:一为TSHR基因突变致TSHR功能异常;另一种为受体后(如cAMP生成)障碍。TSHR基因突变的类型多达20多种。Sunthornthepvarakul等对3例患者及其父母的TSHR基因测序证实,患者的外显子6有两种突变。一个等位基因(T→C)突变导致TSHR的I162N突变;另一个等位基因为C→A突变(P162A)。甲状腺发育不良,血TSH显著升高,T3、T4下降。两种突变型TSHR基因分别由其父母遗传而来,使患者成为复合性杂合子,TSHR有严重功能缺陷。但由于分泌TSH增多,使TSHR缺陷得到代偿,因此可以缺乏甲减的临床表现。Nagashima等报道1对TSH不敏感的日本同胞兄弟,其TSHR基因存在两个新的复合性杂合子突变。血TSH升高,甲状腺位置和甲状腺激素正常,腺体轻度增生。
不同基因型一般具有不同的表型,如I167N、P162A和R109Q突变者TSH升高,T4正常;儿童纯合子A553T突变者甲状腺发育不全,伴甲低;受体基因错义突变导致结合TSH减少或完全不能结合,缺乏转导信号。但也有可能具有相同表型,如PAX8和TSHR突变患者的甲状腺表型无法区分。无义突变(如W546X)导致受体截短,纯合子无义突变使患者TSH升高,T4降低。
TSH受体后缺陷
Mediiros-Neto 等报道,先天性甲减是由于甲状腺细胞第二信使的cAMP生成障碍,甲状腺球蛋白生成减少。Hager-Malecka等报道的1例42个月龄男孩,身材矮小,发育迟滞,伴皮肤干燥、口渴、多尿、隐睾及佝偻病,血GH、胰岛素、T4、TSH、PTH等正常或升高,用GH、T3、T4、维生素D3等治疗后,血和尿中cAMP无升高,这可能是由于腺苷环化酶系统缺陷而导致了包括TSHR后不敏感综合征在内的多激素(TSH、GH、PTH、胰岛素和儿茶酚胺等)不敏感综合征。Congdon等报道,散发性先天性甲减合并甲状腺增生不良病例存在PAX8转录因子基因的杂合子突变(如A119C和Q40P等),其中1例患者的母亲也有同样突变,而且合并慢性淋巴细胞性甲状腺炎。
其他相关性缺陷
主要有:①甲状腺转录因子1 (TTF1)基因突变;②TSH基因突变(分子量增大而无生物活性);③Down综合征患儿常伴有先天性甲减;④TSHR阻滞型抗体(TBAb)导致胎儿一过性先天性甲减和甲状腺发育延迟。
临床表现组成从无症状到严重甲减的连续症状谱
本综合征的症状特点为:
- 临床表现不均一,可从无任何症状到症状严重的甲减,但是甲状腺的位置及大小正常。
- 缺陷严重者有甲减的临床表现,如生长发育、骨骼发育、智力发育延迟等。体格检查有甲减的体征,如黏液性水肿、身材矮小、智商比同龄正常儿童低、牙齿和骨骼发育延迟和跟腱反射恢复期时间延长等。血T3、T4和甲状腺摄碘减低,血TSH明显升高。注射TSH后甲状腺无反应或反应减低。
- 部分病例的父母为表亲婚配(杂合子,多无临床表现)。
- TSHR结构或功能异常,偶尔合并遗传性糖皮质激素缺乏症,部分病例的TSHR正常。
- 病理改变有很大差异,可能与缺陷的严重性有关;甲状腺中度纤维组织增生,滤泡腔胶质减少(甲状腺球蛋白生成减少)。
根据TSH明显升高/甲状腺对TSH无反应/突变基因分析确立诊断
具有下列情况之一者,应怀疑本综合征可能:①甲减表现伴血T4、T3降低而甲状腺不肿大,且甲状腺位置正常;②家族中有已确诊的本综合征患者;③TRH兴奋试验TSH有过分反应,但无血T3/T4升高反应;④先天性甲减;⑤G蛋白病(如假性甲旁减或假-假性甲旁减)。
Takamatsu等提出的临床诊断标准为:①甲状腺位置正常;②甲状腺大小正常或萎缩;③TSH明显增高并具有生物活性;④甲状腺对TSH的反应性降低。血T3、T4和Tg降低。
肯定病因应做有关分子生物学检查。确诊应证明患者的TSH具有生物活性。可克隆分离TSHβ亚基基因,进行cDNA测序并与正常的β亚基基因比较,由正常的TSHβ亚基基因所表达的TSH具有生物活性。有的学者还发现患者血甲状腺球蛋白减少或不可检出,注射TSH后不增高。此外,还应测定甲状腺自身抗体,对患者家系作调查。TSHR基因突变类型很多,可对照文献进行TSHR基因的筛查与鉴定。经甲状腺细针穿刺,获取甲状腺组织,提取细胞DNA可作TSHR基因或Gsα基因的突变分析。现已制备出抗TRH受体的抗血清,可用Western杂交和激光共聚焦显微镜来检测和鉴定TRH受体,协助TRH受体突变的诊断。
TSH不敏感综合征与多种甲状腺疾病鉴别
先天性甲状腺不发育/异位甲状腺
患者在出生后即有甲状腺肿大,甲状腺摄碘率减低,过氯酸钾排泌试验阳性。血Tg不降低,有的患者伴有耳聋。根据酶缺陷的种类和严重程度不同,临床上有不同程度的甲减,代偿完全者可无甲减的表现。
获得性甲减
本综合征应与其他原因引起的先天性甲减和一些后天性甲减进行鉴别(下表),其中变异型TSH甲减是由于TSH结构变异使TSH生物活性减低,如TSH-β亚基的纯合子突变(C105V)引起的遗传性甲减于出生后即有典型甲减表现。TSH不敏感综合征者的TSH活性则正常,可将FRTL-5细胞与提取的TSH一同温育以测定TSH的生物活性。
TSH不敏感综合征与其他先天性甲减表型的鉴别
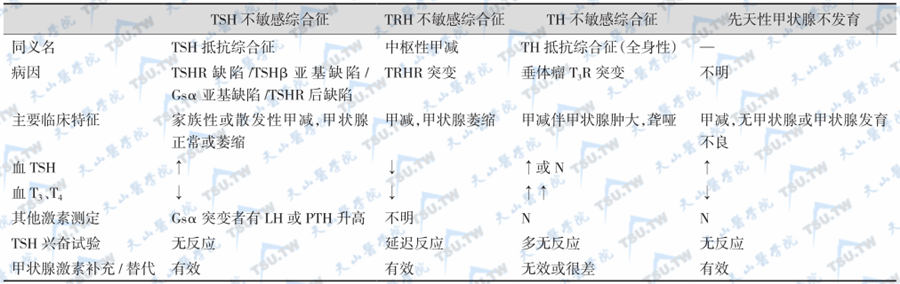
注:TH:甲状腺激素;TSHR:TSH受体;T3R:T3受体;↑:升高;↓:降低;N:正常
慢性淋巴细胞性甲状腺炎
是引起原发性甲减的常见病因。根据本病有甲状腺肿大,血TgAb和TPOAb阳性及甲状腺活检有大量淋巴细胞浸润及滤泡中Tg不减少,可与本综合征鉴别,但慢性自身免疫性甲状腺炎患者对TSH的反应有代偿性抵抗,桥本甲状腺炎可伴有TSHR基因突变。此外,桥本甲状腺炎还可合并转录因子PAX8基因突变,引起先天性甲减。
全身型甲状腺激素不敏感综合征
由于垂体和周围靶器官对甲状腺激素作用不敏感,大多数病例是由于甲状腺激素受体缺陷所致,其共同的临床表现为甲状腺肿大、聋哑、甲状腺功能减退或正常,但T3、T4升高。目前已有600多例甲状腺激素不敏感的病例报告,绝大多数是由于T3R基因缺陷所致,突变部位主要位于T3R基因的C端外显子,呈常染色体显性遗传。TSH正常或升高。外源性甲状腺激素制剂不能纠正甲减。过量甲状腺激素不引起甲亢表现。但应注意,中枢性甲状腺激素不敏感综合征由于下丘脑-垂体对循环T3、T4不敏感,而外周组织对T3、T4的敏感性正常,故可导致甲亢。
TRH不敏感综合征
患者表现为中枢性甲减,血T3、T4、TSH均降低,但与垂体TSH缺乏所致的垂体性甲减不同的是,TRH兴奋试验后,不但TSH对TRH无反应,而且PRL亦无反应(单纯性TSH细胞缺乏者应有反应)。
甲状腺激素补充/替代治疗有部分效果
此综合征患者的治疗取决于甲减的严重程度。对在临床上无甲减症状,发育正常,血T3、T4正常,只有血TSH增高的患者,一般用L-T4补充/替代治疗,满意的补充/替代治疗的目标是血TSH、T3和T4浓度在正常范围。如果TSHR缺陷严重,出生后又未得到及时治疗,可使患者的身体和智力发育出现严重障碍,以后即使给予甲状腺激素制剂治疗,效果也不满意。
甲亢时,可用右旋-T4(D-T4)或3,3,5-三碘甲腺原氨酸(3,3,5 triiodo-thyroacetic acid,RIAC)治疗。
