
先天性高胰岛素血症是β细胞胰岛素分泌调节紊乱引起的一种临床综合征,主要见于新生儿和婴幼儿,称为婴幼儿持续性高胰岛素血症性低血糖症(PHHI)或家族性高胰岛素血症和胰岛细胞增生(familial hyperinsulinism of pancreatic nesidioblastosis),是婴幼儿低血糖症的较常见病因。目前已知,至少有7个基因(ABCC8、KCNJ11、GCK、GLUD1、HNF4A、HADH和SLC16A1)突变可引起PHHI,大约占全部PHHI病例的50%。在组织学上,PHHI分为弥漫性、局灶性和非典型性β细胞增生。弥漫性增生为常染色体隐性或显性遗传,而局灶性为散发性发病;前者需要行胰腺近全切除术,而后者仅需局部切除即可。
PHHI分为多种类型
磺脲类受体及内向调校钾通道相关的PHHI
胰岛β细胞膜上ATP敏感的K+(KATP)通道突变使β细胞去极化,继而电压敏感的钙离子通道开放,钙离子内流,K+通道活性丧失,膜电位活性与糖代谢失耦联,导致胰岛素持续分泌。目前认为,PHHI与胰岛β细胞ATP敏感钾通道的两个亚单位,即磺脲类受体SUR1(ABCC8)及内向校正钾通道Kir6.2的基因突变有关。SUR1/Kir6.2基因位于11p,SUR1的突变热点在第4号外显子(V187D)。β细胞的K+通道调节膜兴奋性与葡萄糖-胰岛素分泌耦联分子由SUR1和Kir6.2蛋白组成,Kir6.2组成K+通道的孔样结构(四聚体),因此,正常的K+通道分子式是(SUR1/Kir6.2)4,K+通道为维持β细胞静息膜电位所必需。
腺苷单核苷酸调节K+通道的活性,因此SUR1受体也是感受核苷酸变化的受体,SUR1可修饰Kir6.2与ATP的亲和力,ATP抑制K+通道活性而ADP拮抗ATP的抑制作用,ADP减少(葡萄糖分解时)导致K+通道关闭。SUR1和Kir6.2的突变类型很多(下表),常见类型有H125Q、 N118S、F591L、T1139M、R1215Q、G1382S、R1394H和F1388等(下表),多呈常染色体显性遗传。这些突变类型的共同特点是SUR1对Mg2+-ADP兴奋性反应下降。临床上低血糖表现最严重,内科治疗无效,患者需行胰腺次全切除。
人SUR1基因变异
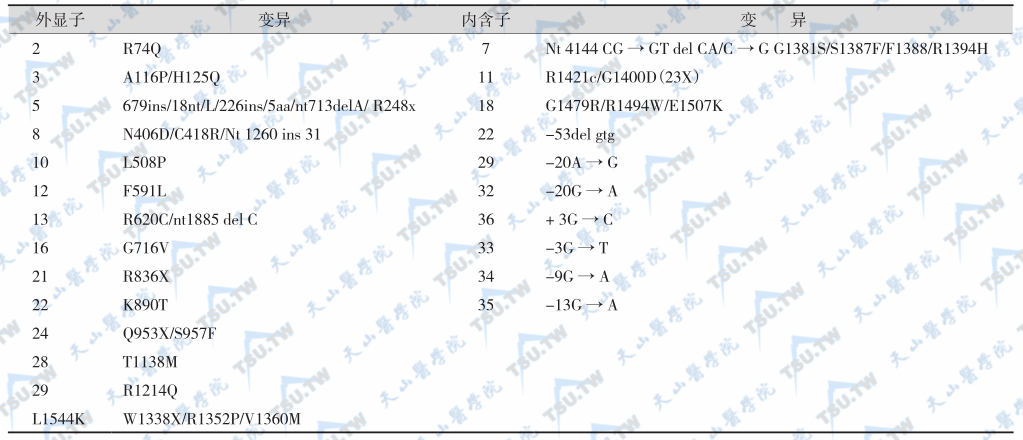
导致先天性高胰岛素血症的致病基因
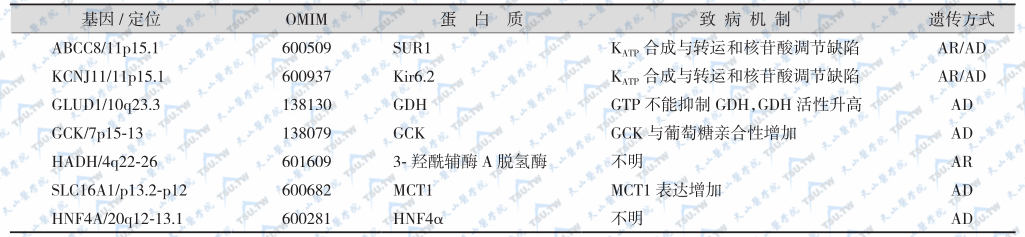
注:SUR1:sulfonylurea receptor 1,璜脲受体1;Kir6.2:Inward rectifying potassium channel,内向调校性钾通道;GDH:glutamate dehydrogenase,谷氨酸脱氢酶;GCK:glucokinase,葡萄糖激酶;MCT1:monocarboxylate transporter 1,单羧-转运体1;HNF4α:hepatocyte nuclear factor4α,肝细胞核因子4α;AD:autosomal dominant,常染色体显性遗传;AR:autosomal recessive,常染色体隐性遗传。
谷氨酸脱氢酶相关的PHHI
谷氨酸脱氢酶(glutamate dehydrogenase,GDH)突变是仅次于K+通道突变的PHHI类型,常呈常染色体显性遗传。GDH催化谷氨酸的氧化脱氨过程,产生α-酮戊二酸和氨,而α-酮戊二酸作为三羧酸循环的底物与糖代谢密切相关。GDH活化性突变导致α-酮戊二酸及ATP/ADP比值升高而与高胰岛素血症性低血糖症及高氨血症相关,通常对二氮嗪治疗的反应良好。血氨可升高3~5倍,但患者通常没有高氨血症相关的症状。
谷氨酸盐(glutamate)除了作为特殊的神经递质与信号分子外,还在细胞内信号传递中起了重要作用。在肝脏、肾脏、脑组织和胰腺胰岛中,谷氨酸盐通过线粒体基质的谷氨酸脱氢酶(glutamate dehydrogenase,GDH)催化其脱氢氧化为α-酮戊二酸(alpha-ketoglutarate)。GDH的活性调节十分复杂,包括了负性(如GTP和乙酰辅酶)和正性(如ADP与亮氨酸)两个方面。在一定条件下,GDH活化性突变(ABCC8、KCNJ11、GLUD1、CGK、HADH、SLC16A1和HNF4A)导致GTP对GDH的负性抑制作用丢失,故引起儿童高胰岛素血症-高氨血症-低血糖综合征(hyperinsulinismhyperammonemia-hypoglycemia syndrome)。本综合征的低血糖发作自幼开始,短暂禁食或进食蛋白餐为常见诱因,发作时血糖明显降低而血氨升高3~5倍。
葡萄糖激酶相关的PHHI
葡萄糖激酶(glucokinase,GCK)不仅是β细胞内葡萄糖代谢的限速酶,而且是β细胞的“葡萄糖感受器”,介导葡萄糖刺激的胰岛素分泌。GCK活化性突变的结果是葡萄糖刺激的胰岛素分泌阈值“重设”,多表现为轻中度的PHHI,同时表现为空腹和餐后低血糖,血胰岛素水平轻中度升高,一般对药物治疗反应良好。
由于突变方式不同,葡萄糖激酶突变后可分别引起高血糖症或低血糖症。一般来说,GCK的杂合子失活性突变导致成年起病的青少年糖尿病(maturity-onset diabetes of the young,MODY),其特点是自幼发生的轻度空腹高血糖,但往往不能被早期诊断。纯合子失活性突变则引起自幼起病的永久性新生儿糖尿病(permanent neonatal diabetes mellitus,PNDM)。另一方面,许多GCK的杂合子活化性突变(heterozygous activating GCK mutations)也引起低血糖症。目前,已经报道了1441个家族的620个GCK突变位点,其中多数的活化性突变位于所谓的变构激活物部位(allosteric activator site)。
其他原因所致的PHHI
有些学者把短链3-羟酰-辅酶A脱氢酶(SCHAD)基因(SCHAD-HI)和磷酸甘露糖异构酶缺陷症也归为PHHI的亚型之一。严格来说,SCHADHI属于线粒体呼吸链疾病(mitochondrial respiratory chain disorders,MRCD)的范畴,是引起儿童肝衰竭的主要病因之一,患者可表现为低酮症性低血糖和乳酸性酸中毒。SCHAD 是MRCD的一种临床亚型,呈常染色体隐性遗传。患者脂肪氧化障碍,故容易发生低血糖症,但不伴或仅伴有轻度酮症。磷酸甘露糖异构酶缺乏症属于蛋白糖化酶系突变引起的先天性代谢病(碳水化合物缺乏性糖蛋白综合征,糖蛋白碳水化合物缺陷综合征,CDGS)1b型,以血清多种糖蛋白(特别是血清转铁蛋白)异常糖化和糖化障碍为标志。由于磷酸甘露糖异构酶缺乏,导致6-磷酸果糖向6-磷酸甘露糖的转变发生障碍。患者表现为低血糖症、蛋白丢失性肠病和肝损害,心脏、肾脏、肌肉和神经系统亦可受累。一般可以用甘露糖治疗。
原因未明的PHHI
约50%的患者经上述突变基因的筛查未发现异常;多数经二氮嗪治疗有效,提示患者存在K+通道异常,但至今仍找不到确切的变异分子。有些患者的临床表现呈一过性,低血糖症在数月内可以自行缓解;有的呈丙酮酸可诱导性低血糖症。
从婴幼儿高胰岛素血症性低血糖症病例中筛查和诊断PHHI
18F-DOPA-PET有助于诊断。但确诊有赖于组织病理学检查和突变基因分析。PHHI的一般诊断依据是任何时间的高胰岛素血症(多数≥1000μU/ml)伴低血糖(<3.0或2.8mmol/L),或胰岛素/血糖比值>0.3,或胰高血糖素阳性。年龄多在2岁内,多数伴有高氨血症(hyperammonemia)并排除了其他原因引起的高胰岛素血症和低血糖症。先天性高胰岛素血症病因鉴别的要点是相关基因突变分析。对于儿童和青少年患者,如果排除了后天性高胰岛素血症性低血糖症的可能,那么就要考虑先天性高胰岛素血症的诊断,并通过进一步检查明确病因。PHHI主要见于婴幼儿和新生儿,当可疑对象有低血糖症家族史、持续性血胰岛素水平升高和反复发作性低血糖症,而胰腺影像检查阴性时,要注意PHHI可能,可行18F-L3,4-二羟苯丙氨酸-PET(fluorine-18 L-3,4-dihydroxyphenylalanin)检查。
高胰岛素血症亦可见于Beckwith-Wiedemann综合征、Soto综合征、Costello综合征、Timothy-Kabuki综合征和先天性糖化病(congenital disorders of glycosylation,CDG),应注意鉴别。
用甲醛溶液固定,石蜡包埋的胰腺组织切片,观察到两种PHHI类型(下表)。
PHHI病例的不同特征
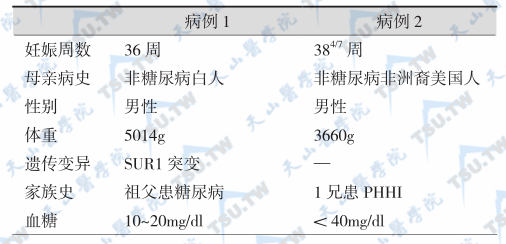
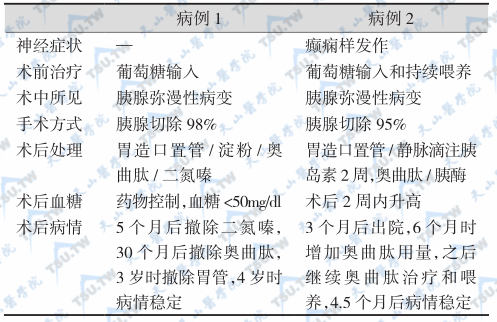
注:PHHI:persistent hyperinsulinemic hypoglycemia of infancy,婴幼儿持续性高胰岛素血症型低血糖症;SUR1:sulfonylurea receptor-1,璜脲受体-1。
根据情况选用对症治疗或手术治疗
绝大多数PHHI不可能治愈(局灶性β细胞增生可手术治愈),只能用药物控制症状,而手术治疗仍无统一意见。有人主张切除胰腺95%以上,但易于发生糖尿病。胰岛细胞增生症(nesidioblastosis)可能为局限性病变,因而可做局部切除术,增生型或混合增生型可做全胰切除术,胰腺次全切除可能更适合于弥漫性增生型患者。
PHHI的治疗主要为对症性的,新生儿和婴幼儿要防止低血糖反复发作,以防发生不可逆性脑损害。PHHI引起的低血糖症死亡率高,治疗困难,药物治疗如二氮嗪(同胰岛素瘤)、奥曲肽及钙通道拮抗剂可能有助于控制低血糖症。手术被认为是一种有可能治愈本病的手段,但必须尽早实施。目前认为,PHHI有两种组织病理学类型:局灶性胰岛细胞腺瘤样增生和弥漫性β细胞功能亢进,对两者可分别施行胰腺部分切除术及胰腺次全切除术,Sempoux等报道,患有PHHI的新生儿中前者占40%,而且预后明显比后者好,及早诊断与手术治疗有助于改善预后。McAndrew等报道,手术的并发症有术中出血,损伤脾、胆道和小肠等,部分患儿因术后胆道漏及胆道狭窄需行胆总管十二指肠吻合术,另一些患儿术后需用胰岛素和胰酶替代治疗。如果低血糖仍反复发作,则用胰高血糖素加低剂量奥曲肽治疗。(廖二元)
