
骨-软骨增殖不良症伴有特殊表现
在临床上,应仔细测量臂距、坐高、上身高(顶-耻距)、下身高(耻-跟距)及头围,并确定长骨、脊椎骨和颅骨等是否有畸形及其他异常。在X线片上,还要根据临床表现确定骨骼异常的原发部位在骨干、骨骺、干骺端、骺软骨或软骨。以软骨无增殖症(achondroplasia)较常见。软骨无增殖症为常染色体显性遗传病,但90%的病例为新突变者。例如,成纤维生长因子受体-3(基因位于4p16.3)的穿膜区突变(基因的突变热点为1138),可导致软骨无增殖症。
纯合子患者于出生后因胸廓狭小,肺功能不全难以存活。幸存者生长缓慢,平均高度约为120cm(女性)或130cm(男性),较年长患者常具有特征性骨骼畸形表现。头颅大、鼻梁低而平、前额突出和上颌小,但躯体长度基本正常。肢体短,顶-耻距明显大于耻-跟距。双手下垂仅及髂部,软组织增生过度,皮肤折叠明显,肢短和步态蹒跚。可伴有脊髓或神经根受压症状,部分患者有脑积水及颅内高压症状。全部病例有腰椎前突、臀部后翘、肘关节屈曲及伸展受限。约半数病例有婴儿期腰椎后凸,约1/5病例有上腰椎后凸。有时可见脊柱侧弯、髋或膝内翻。一般腓骨的干骺软骨较少受累,而使腓骨长于胫骨。X线可发现面颅小,枕骨大孔狭小可致脑积水。
颅底短,脊柱的椎弓缩短,椎体后缘呈扇形凹陷,椎弓根间距自上至下逐渐变短,其连线呈漏斗状(正常人为“八”字形),上部腰椎椎体可呈楔形变,持续性腰椎后突使骶骨后翘。胸廓短小,呈扁平胸,髂骨翼小而呈方状,髋-臼顶呈水平位,三角软骨内缘可有一尖刺样骨突。坐骨大切迹狭小,股骨近侧干骺端增宽和外展,股骨颈增宽和变短,颈干角变小。骨骺常正常,管状骨短而粗,干骺短而增宽,并呈喇叭口样外展,骨干可弯曲,腓骨较胫骨长。双手呈“海星”或“三叉”状,短管骨变短,掌骨等长。
根据影像检查和突变基因分析确立诊断
如患者成年,多数健壮。部分有下肢瘫痪或驼背。由于腓骨过长,易引起膝和踝关节负重不均匀而导致骨关节病。本病应与软骨发育减退症和假性软骨发育不全等鉴别。干骺端软骨发育异常(metaphyseal chondrodysplasia)应与佝偻病鉴别。软骨发育不良症(chondrodysplasia)并非软骨无增生症的亚型,也非同一基因突变所致。本症无面部畸形,临床上通常表现为短肢型身材矮小,手臂短,顶-耻距大于耻-跟距,可伴腰椎前突,肘关节伸展受限及膝内翻。成年身高约120~150cm。
1型PTH/PTHrP受体突变可导致软骨发育不良症。Jansen软骨发育异常是一种活化型受体突变,患者出现高钙血症和骨生长异常。父源性印迹基因(imprinted genes)的特异性表达影响子代的生长发育。因此许多遗传性印迹疾病(congenital imprinting disorders,IDs)的重要临床特点之一是生长障碍。Silver-Russell综合征的特点是宫内和出生后生长障碍、巨脑和三角脸等。患者的7号染色体均为母源性(10%),11p15印迹控制区低甲基化(38%)或染色体畸变(1%)。
Ras/有丝分裂原(mitogen)-活化蛋白激酶(mitogen activated protein kinase,MAPK)途径主要调节细胞周期、分化、生长、发育和衰老。由于MAPK途径调节异常所致的发育障碍统称为RAS病(RASopathies),这些疾病的共同特点是Ras/ MAPK途径中的因子出现了胚系突变(germline mutations),该类疾病很多,虽然各有特征,但因涉及的信号途径相同,因而其临床表现往往重叠,如面部与心血管畸形、皮肤异常和神经精神障碍等。
Ras/MAPK途径因子突变所致的遗传综合征
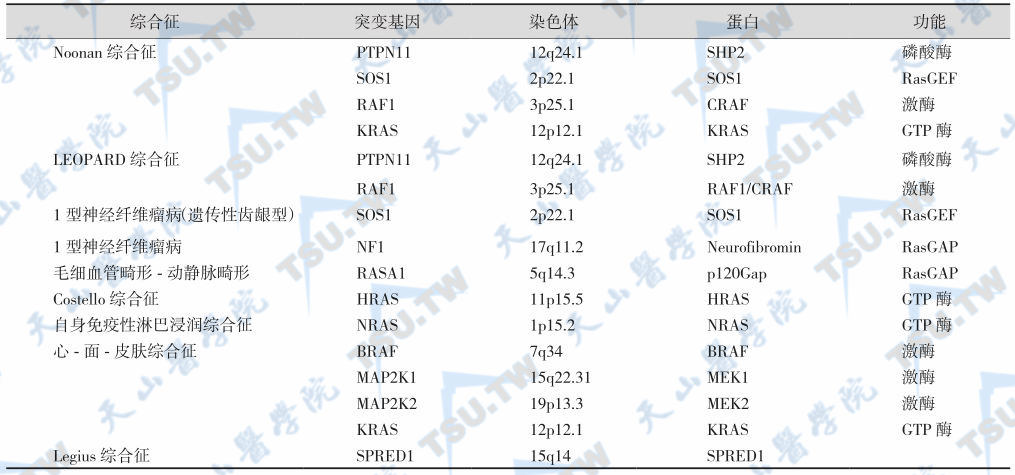
注:1型神经纤维瘤病:neurofibromatosis type 1;1型神经纤维瘤病遗传性齿龈型:congenital gingival fibromatosis 1;毛细血管畸形-动静脉畸形:capillary malformation-arteriovenous malformation;自身免疫性淋巴浸润综合征:autoimmune lymphoproliferative syndrome;心-面-皮肤综合征:cardio-facio-cutaneoussyndrome
Noonan 综合征
PTPN11或KRAS基因突变导致Noonan综合征,呈常染色体显性遗传,偶呈常染色体隐性遗传。其特点是身材矮小、面部畸形(倒三角脸)和先天性心血管-胸廓畸形、前臂外翻、关节过度伸展、指(趾)短小、平板足/马蹄内翻足/畸形足、桡-尺骨融合、下颌骨粗大(巨细胞损害所致)、性腺功能异常、出血素质与恶性肿瘤、淋巴管发育不良症/不发育症(lymphatic vessel dysplasia/hypoplasia/or aplasia)、色素沉着异常、斜视、弱视、黄斑异常(20%)、眼球震颤、听力缺失和肝脾肿大等。
LEOPARD综合征
常染色体显性遗传,临床表现与Noonan综合征相似,如Noonan样面形、多发性色素痣、心电图异常、眼距过宽、肺动脉瓣狭窄、生长迟缓和耳聋等。
1型神经纤维瘤病与神经纤维瘤病(齿龈型)
呈常染色体显性或隐性遗传。1型神经纤维瘤病(NF-1)是由于广泛多样的基因突变所致,尤其是等位基因丢失引起神经纤维素活化,GTP酶的功能缺失导致p21ras活化。根据病理特征与伴发疾病(如嗜铬细胞瘤、癌瘤、甲状腺髓样癌、原发性甲旁亢、青春期异常和性分化异常),NF可分为六型。主要的病理特征为分布于脊神经、脑神经、皮下或皮下神经的多发性神经纤维瘤以及因表皮基底细胞层内黑色素沉积而致皮肤色素斑。神经纤维瘤病(齿龈型)病情进展较慢,临床经过为良性,齿龈过度角质化。1型神经纤维瘤病(齿龈型)是由于SOS1基因插入性突变所致。
毛细血管畸形-动静脉畸形
呈常染色体显性遗传。毛细血管畸形为多灶性,可发生于许多组织,如皮肤、肌肉、骨骼、心脏、肺脏或毛组织,常伴有动静脉瘘。
Costello综合征
主要表现为发育障碍,如颅面不对称型畸形,心血管和神经系统功能异常以及生长迟缓等,常合并各种肿瘤。
自身免疫性淋巴浸润综合征
其特点是淋巴细胞凋亡障碍,引起非恶性的淋巴细胞大量积聚,因而最终导致血液系统肿瘤。
心-面-皮肤综合征
临床表现与Noonan综合征相似,但神经系统的异常更常见而严重,如肌张力低下、动作迟缓和语言障碍等。
Legius综合征
临床表现与神经纤维瘤病相似,可有皮肤café-au-lait斑、轻度神经功能障碍及面部畸形等。
