
英文:Marfan syndrome、MS(MIM 154700);
同义名:马方综合征、马凡氏综合征、蜘蛛指(趾)病、细长肢体病、肢体细长病、长肢病、指趾过长综合征、长瘦忧郁症、蜘蛛手合并晶体移位。
溯源与发展
1896年巴黎儿科医生Antoine Marfan首次报道1例5岁女孩,有显著的骨骼异常,他称为细长肢体病。1902年Achard重新命名为蜘蛛指(趾)病。1914年Bogei首先将晶体脱位与蜘蛛指联系起来。1931年Were综合84例命名为Marfan综合征(Marfan syndrom,MS),首次指出本病遗传方式为AD。1943年Baer等报道患有主动脉扩张和夹层动脉瘤,并指出本综合征也见于成人。1955年McKusick报道和分析50个家系105名患者的骨骼、眼及心血管损害,强调为遗传性疾病。
国内早在1915年起已采用首报者马方的名字而命名为Marfan综合征,另外尚有马方综合征、马凡氏综合征、蜘蛛手合并晶体移位等多种名称。1983年遗传学家杜传书等采用蜘蛛指(趾)综合征的命名,同年梁国芬及其后孙启斌、张开滋等进行了临床和遗传学研究,并建议国内应统一采用Marfan综合征为妥。其特点为外显率高,表现度不一,主要累及骨骼、眼及心血管系统的结缔组织,呈AD方式为特征的综合征。
发病机制
本病系一种纤维结缔组织遗传病,病因是原纤维蛋白基因突变导致全身中胚叶组织广泛发育不良,而引起多系统疾病。
遗传学:主要表现在以下几个方面:
- 流行病学:孙启斌、张开滋、郑宗锷等回顾性地收集了中国1951-1988年所报道的610例Marfan综合征,同时也普查了29067名儿童,其结果示群体患病率为0.72%,基因频率为8.61x10-4,外显率为71.28%,突变率为3.09x10-5。这与陆炳新的报道相似。
- 遗传方式:McKuaick报道50个家系105例马方综合征中,AD占85%,Cross和Jonson报道AR占1.08%;与西方国家相比较:
- 我国有家族史者为455例占74.59%,其中AD占73.6%,AR占0.98%;
- 散发病例为155例占25.4%,这可能由于基因突变或前代个体所携带致病基因未外显或家系调查不完全所致。
- 基因定位于15q21.1;非典型为7q22.1。25%~30%为新发生的基因突变。
- 表现度有较大差异:Marfan综合征在临床上可有骨骼、眼、心血管均改变的完全型,亦可表现为分别由上述组合的不完全型。即使在同一家系中其表现亦有多少或轻重之分。这种表型的不一,是基因多效性所致。
- 皮纹改变:
- 斗形纹略增多,常出现在第1、4指上。
- 向心位轴三叉点,且向桡侧移位,致使atd角缩小,t-桡距离缩短。
- 手掌呈屈褶纹,为垂柳状分布,垂向手掌的向心部。
临床表现
骨骼病变
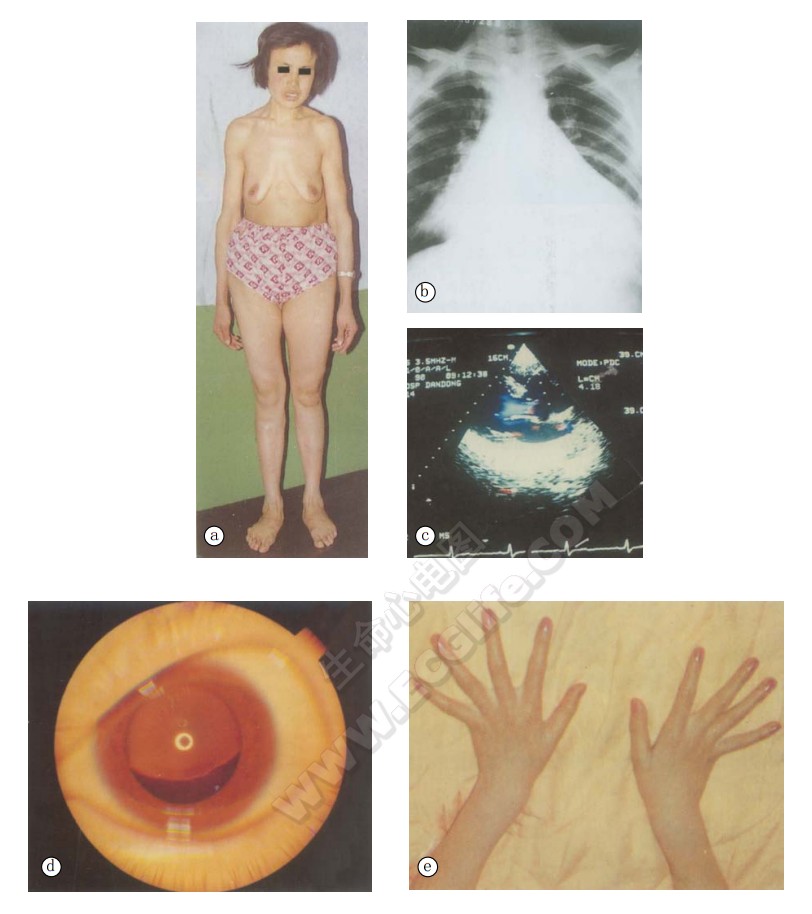
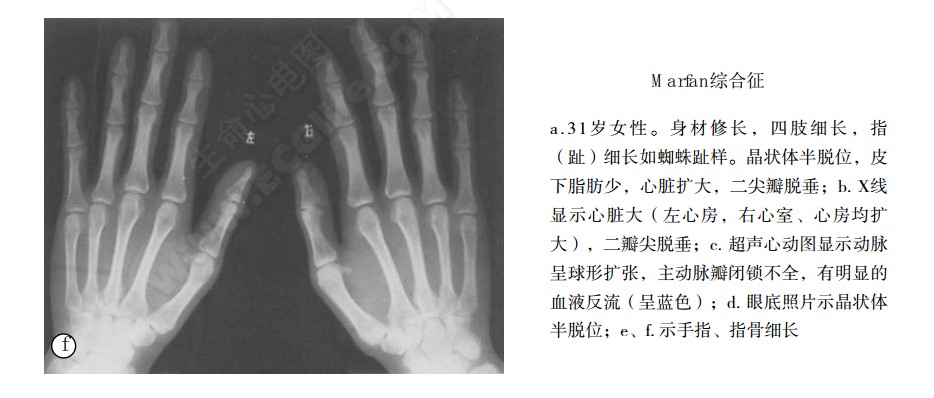
可累及许多部位骨骼并呈不同程度表现,我国610例骨骼受损率为97.2%,与国外文献报道相近。Marfan综合征的身材瘦高和蜘蛛指(趾),为最常见的骨骼异常。其出现率分别为84.4%和77%,与国外报道的80%无明显差异。Marfan综合征者身材瘦高,四肢细长,特别是前臂和大腿更为明显,指距大于身长,上半身比下半身短,其比值<0.92。手指和脚趾特别长,呈蜘蛛样指(趾);手与身的之比>11%;脚与身高之比>15%。由于身体各部位纵向生长过度,因而发生头长、窄面、高腭、凸颌、鸡胸、漏斗胸、驼背、脊柱侧凸和扁平足等改变。在骨骼畸形中以非对称性鸡胸、漏斗胸较有意义,蜘蛛指较有重大特异性。另外还有皮下脂肪少,肌肉发育不全,关节松弛,易患腹股沟疝。
腕征(患者以一手拇指和小指握住对侧手腕桡骨茎头近端处,如果拇指和小指相互重叠,则为阳性,它反映手指长和手腕细二者比例发生改变)和拇指征(患者拇指内收,横置掌心,伸直并握拳,如果拇指超出该手尺侧缘,则为阳性)对诊断有所帮助,笔者检查41例,两者出现率分别为65.9%和85.3%,国外报道两者分别为58%和82%,值得注意的是有假阳性和假阴性。Siachain等介绍正常人掌骨指数(双手X线后前位片上,将食、中、无名和小指的4个掌骨长度与4个掌骨中部的平均宽度之比值)<8,Marfan综合征>8在8.4与10.4之间,(男性>8.4、女性>9.2),笔者检查率为80%。Suridge指出掌骨指数特异性远远超过指距/身长和上半身/下半身之比率,笔者同意此意见并认为较腕征和拇指征可靠性更高。
眼部病变
笔者资料眼部病变出现率为63.8%,其中晶体脱位为最常见病变,出现率为86.8%,与西方国家报道83%相近。晶体的半脱位、全脱位导致继发性眼部病变及合并轴性屈光异常,导致近视,并发视网膜剥离。还易患虹膜震颤、白内障等病。
心血管病变
可早至5岁,晚至60岁,常为进行性。我国的Marfan综合征心血管病变发现率为38.5%,西方文献报道为40%~60%,但病理检查心血管损害发生率为95%~100%。主要是主动脉中层坏死。川岛综合日本文献77%病例有主动脉中层坏死以及二尖瓣黏液变性。主动脉病变包括升主动脉扩张形成升主动脉瘤,以及主动脉扩张伴主动脉瓣关闭不全。Sllis等称之为“主动脉环呈动脉扩张症”,为Marfan综合征的特征性改变。这种扩张最早和最多发生在主动脉根部。主动脉瓣关闭不全并心衰及主动脉瘤或夹层动脉瘤破裂是主要死因。
冠状动脉受累而发生心绞痛、二尖瓣黏液变性使瓣叶变薄、过长或腱索伸展导致二尖瓣脱垂(MVP)。Brown等提出超声心动图诊断主动脉扩张的标准为:
- 主动脉宽度>22mm/m2;
- 实测主动脉内径>37mm;
- 左房/主动脉内径<0.7。
以上三项中具备两项即可诊断。国内同济医科大学附属协和医院超声心动图资料:
- 主动脉根部内径,成人男性>35mm,女性>34mm;
- 左房庄动脉比率≤0.7;
- 可见二尖瓣脱垂(MVP),以后叶多见;
- 若有主动脉或二尖瓣反流,可出现左室和,或左房增大;
- 应除外高血压病、风心病、冠心病引起主动脉根部增宽,以及梅毒性主动脉瘤。
心血管造影对诊断更有较高价值。超声心动图和磁共振对于早期检出主动脉根部扩张比X线检查更具有优越性,国内资料表明超声诊断率为92.3%,而胸片仅为69%。
在西方国家MVP为最常见的瓣膜疾病,在Marfan综合征中出现率超过80%,国内为14%,MVP伴骨骼畸形与Marfan综合征相似。提示MVP可能是Marfan综合征的顿挫型。另—方面,Glesby和Pyeriz认为Marfan综合征伴MVP,两者的关系有待进一步探讨。
早年文献提及其他先天性心血管畸形占1/3,国内资料仅占2.4%,如:房、室间隔缺损、法洛四联症、动脉导管未闭、肺动脉狭窄等,现已被认为与Marfan综合征无病因上关联。少数患者可并发感染性心内膜炎。
硬脊膜膨出
Pyeritz曾对57例Marfan综合征患者以及与其年龄、性别配对的非Marfan综合征57例做对照研究,发现63%Marfan综合征患者可见腰部椎管增宽现象。而对照者无1例发现。国内结果为66.7%与之类同,说明硬脊膜膨出是Marfan综合征的常见特征。Pyeritz研究证实,硬脊膜膨出的发生与骨骼畸形、晶体脱位以及主动脉损害的严重性之间并无一定关系,且腰骶部硬脊膜膨出多无症状。
硬脊膜膨出发生机制是因MS细胞外基质缺陷,致结缔组织生活力下降,硬脊膜长期持续受到脑脊液搏动性压力影响,特别是直立位时,蛛网膜下腔压力在腰骶部椎管最高,造成腰骶部脊柱的侵蚀,蛛网膜下腔囊肿以及盆腔脊膜膨出。
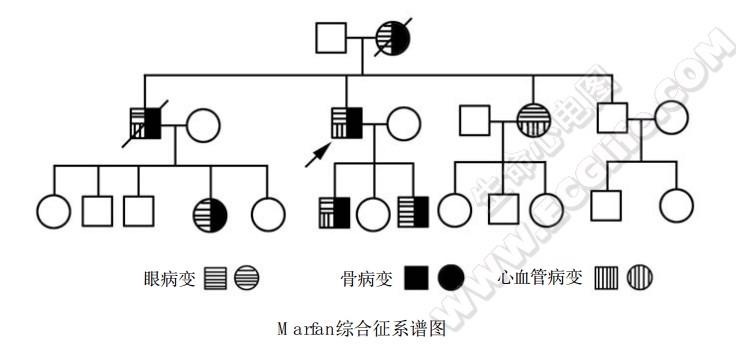
Marfan综合征的临床表现多样,如下图:
诊断
Marfan综合征的诊断依据1964年由Wilnor和Finby提出,尔后1979年由Pyeritz和McKusick修订的四项诊断标准:
- 骨骼病变:蜘蛛指(趾)为最特征性改变;
- 眼部病变:晶体脱位最具特征性;
- 心血管病变:主动脉根部扩张最具特征性;
- 遗传性家族史。
国内资料表明,骨骼异常为最常见,其次为主动脉根部扩张。尿中羟基脯氨酸排泄增加,掌骨指数≥8.4,在诊断上具有重要参考价值。Hirst Core指出主动脉窦扩张为Marfan综合征中膜变性最早的发病部位,可作为诊断线索。笔者认为凡临床上认为不典型风心病的二尖瓣或主动脉瓣关闭不全或青年人呈单纯性主动脉关闭不全,应全面检查,综合分析,注意发现骨骼和眼有无异常,以防漏诊。对瘦高体型,四肢细长怀疑Marfan综合征者如未发现心血管病变,应定期做超声检查,必要时也可做心血管造影以资诊断。总之,具有骨骼、眼、心血管病变和遗传家族史四项中任何两项,即可诊断本病。如仅有心血管病变,而无其他改变者亦应疑及,以防漏诊。1988年国际Marfan综合征会议制定诊断标准见下表。

Marfan综合征分型
- 临床分型:
- 完全型(典型):同时具备骨骼、眼、心血管三主征;
- 不完全型(非典型):只具备三主征中的1-2项。
- 遗传学分型
- 家族性:是上代致病基因遗传所致;
- 散发性:是基因突变,亦可是上代致病基因未外显。
鉴别诊断
需与下列疾病鉴别:
- 同型胱氨酸尿症;
- 家族性或散在性二尖瓣脱垂综合征;
- 家族性或散发性主动脉瓣环扩张;
- 先天性挛缩性蜘蛛样指(趾);
- Marchesani综合征(反Marfan综合征);
- Stricker综合征;
- 18三体综合征。
治疗及预后
预后多数不良。婴儿患者多死于感染:不治疗的患者50%在40岁左右死于心血管病变及并发症,其中死于主动脉瓣关闭不全者占19%,主动脉瘤破裂占16%,夹层动脉瘤占11%。国外文献报道成人92%死于心血管疾病。
目前,尚无满意疗法。对眼部病变可酌情矫正及对症处理。应积极预防治疖心血管并发症和心力衰竭,对主动脉瘤、夹层动脉瘤应争取早期手术治疗。
