
骨的生长发育主要受遗传因素的影响。由于调节骨发育的基因缺陷、变异(染色体畸变或基因突变等)及骨发育障碍,导致各种骨骼畸形综合征,这些疾病或综合征可统称为体质性骨病(constitutional bone disease)。这类疾病的种类繁多、分类混乱,多数疾病的病因未明。近年来,随着分子生物学技术的广泛应用和病因学研究的深入,一些体质性骨病的病因已经查明,有些已经应用于临床诊断或产前诊断,如软骨发育不全综合征、马方综合征等。本节主要介绍五种体质性骨病临床综合征:①身材过高综合征;②骨畸形综合征;③BMD增高综合征;④颅缝早闭综合征;⑤异位钙化与异位骨化综合征。
在内分泌代谢性疾病中,引起身材过高的主要是巨人症与肢端肥大症,其次为体质性身材过高(此病与种族和某些遗传素质有关,但并非病态)。除上述的GH分泌过多和遗传素质外,身材过高主要见于染色体畸变(如47,XXY或47,XYY综合征)和马方综合征与类马方综合征。至于青春发育提前和性早熟所致的身材过长只是相对于年龄而论的,因为其最终身高可正常甚或低于正常。
47,XXY及其变异型表现为男性类无睾体型
上部量明显短于下部量(但上肢一般不过长,指距通常不超过身高)。肌肉发育差,体毛、胡须和阴毛少,常伴男性乳腺发育。46,XY/47,XXY嵌合型的临床表现与累及的细胞数和所在的组织有关。47,XYY患者的社会性别和生殖器性别均为男性,但睾丸及第二性征发育不完善,睾丸细小,常伴乳腺发育。身材高、体力好,可有智力障碍。患者心理行为异常,易导致暴力倾向和行为。48,XXYY型是XYY精子和X卵子或YY精子和XX卵子结合的结果。患者除了具有47,XXY型的临床表现外,还有身材高瘦(平均身高181cm左右)、智能低下、类无睾体型和男性乳腺发育等表现。48,XXXY型的智能低下更明显,体格异常多见,如短颈,内眦赘皮,桡、尺骨融合和指(趾)弯曲等,性腺发育障碍也更明显。
男性化不足可用睾酮替代治疗,男性乳腺发育且心理压力较重者宜行乳腺成形术。
Marfan综合征
Marfan综合征以细长趾和上部量与下部量比降低为特征。Marfan综合征(Marfan syndrome,MFS)病变特征为结缔组织结构缺陷,伴胶原代谢的多种异常(如羟脯氨酸排泄增加),原因在于胶原转化增多,主要与原纤维胶原(fibrillin-1,FBN1)基因突变有关。FBN1由成骨细胞和骨细胞合成分泌,同时这些细胞还表达FBN2及其他微纤维蛋白。FBN1蛋白是构成细胞外微纤维的主要蛋白之一,因此FBN1基因突变将影响微纤维的结构与功能,这类疾病统称为微胶原蛋白病(fibrillinopathies)。临床上分为Ⅰ型微胶原蛋白病和Ⅱ型微胶原蛋白病两型;前者主要累及骨骼、心血管和眼睛,表现为MFS及其相关性疾病。
Marfan综合征的病因是15号染色体上的原纤维蛋白基因突变。眼睛、骨骼及心血管有特征性的临床表现,80%的患者有心脏大血管受累。通常5岁(35%)发生主动脉扩张,到19岁时,68%的患者有临床表现。β受体阻滞剂是经典治疗药物,尽管该药对胎儿有风险(妊娠晚期最高),易发生二尖瓣黏液样变伴脱垂,达到生育年龄时常伴有先天性血管壁中层异常——弹性纤维缺失、平滑肌细胞减少与胶原和基质增多,容易导致动脉瘤、夹层及进行性主动脉瓣反流。妊娠期,血管壁中层异常和血流动力学变化增加这些风险。当主动脉根部直径达到或超过45~50mm,或6~12个月内直径超过2~5mm,或主动脉瓣反流进行性加重时,考虑择期主动脉根部手术及主动脉瓣修复或置换术。主动脉根部直径超过45mm时,建议妊娠前先接受修复性手术;若已怀孕,应终止妊娠。
原纤维蛋白-1基因(FIBRILLIN-1)为该病的候选基因。原纤维蛋白是一种大分子糖蛋白,它与其他蛋白结合后形成微纤维,现已确定了30多种突变类型,因原纤维蛋白-1基因较大(约110kb,含65个外显子),突变筛选较为复杂。TGF-β受体(TGFBR1和TGFBR2)突变引发的综合征与Marfan综合征存在表型重叠现象。Loeys-Dietz综合征的临床特点包括:①动脉迂曲与主动脉瘤;②眼距增宽;③悬雍垂裂或腭裂。血管紧张素Ⅱ受体阻断剂氯沙坦对TGF-β的活性具有拮抗作用。
细长趾约占76%,一般上部量与下部量比(US/LS)低于2个标准差。细长指占88%。其他特征有胸廓畸形(68%),脊椎侧凸(44%),平足与关节囊松弛(44%),关节过度伸展(56%)及复发性脱位(髋、膑、锁骨关节)等。椎体可增大、增宽。
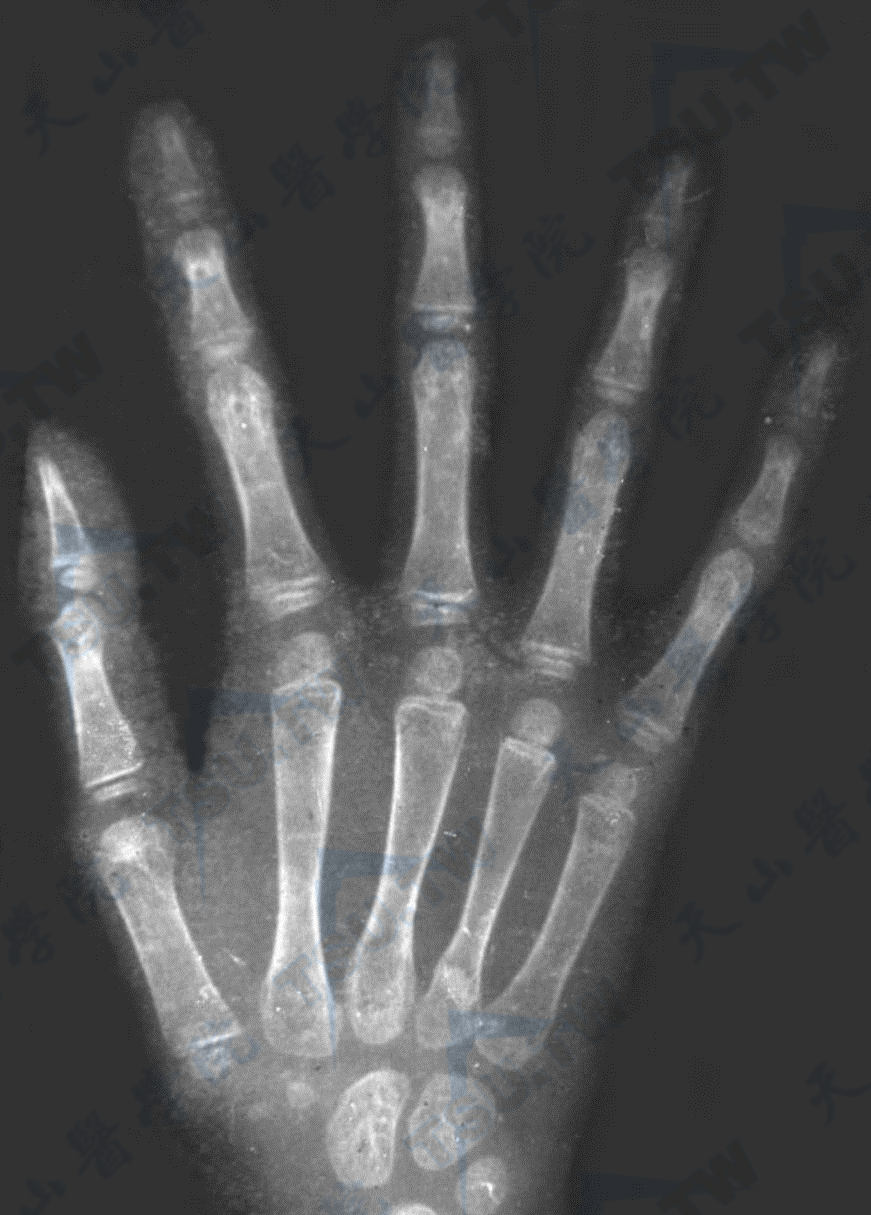
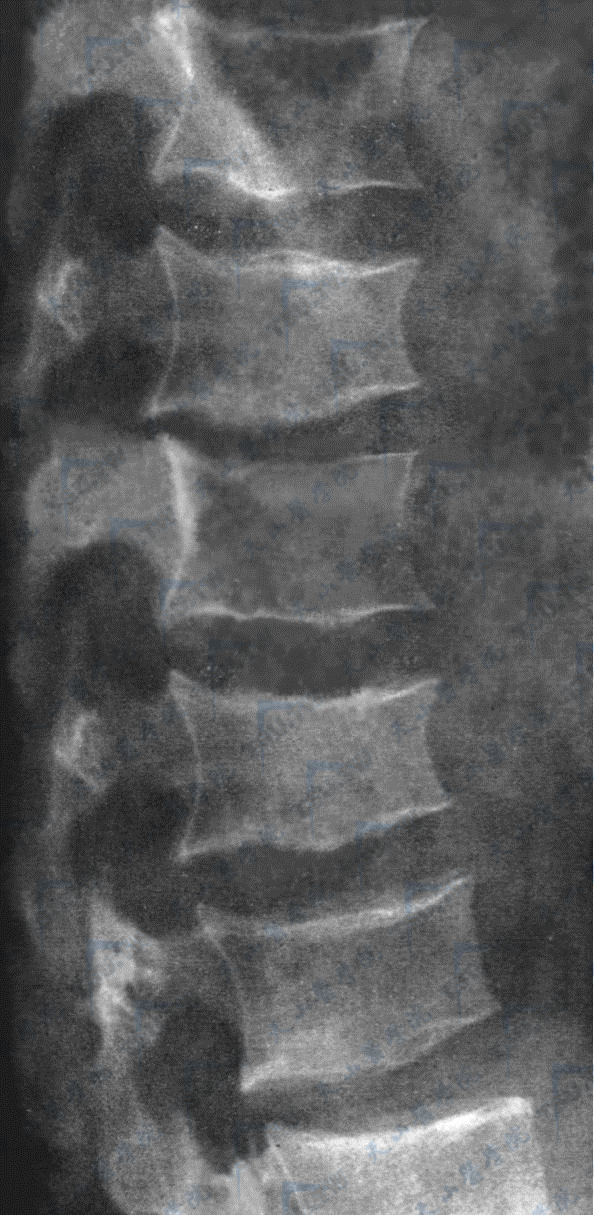
Marfan综合征
注:男,19岁,左手正位片。掌骨及指骨细长(掌骨指数10.3)。胸腰段脊椎侧位像。示椎体前后径稍增长,椎体上下终板欠规则,椎体后缘略呈贝壳样改变。
MFS应与多种疾病鉴别:
- 类Marfan综合征体型。见于胱硫醚合酶缺陷症(高胱氨酸尿)、先天性挛缩性细长指(趾)、MFS样过动综合征、类无鼻症、Klinefelter综合征、XXY综合征、镰刀型贫血、多发内分泌腺瘤综合征(Ⅱb型)及痣样基底细胞癌综合征。Stickler综合征及多发内分泌瘤Ⅰ型部分病例也有MFS样体型。
- Shprintzen-Goldberg综合征罕见,以颅缝早闭和类马方体型为主。根据一些反常面部特征识别,如有细长指(趾)、颅缝早闭、智力缺陷、两眼间距过大、眼部突出、眼睑下斜、小颌、耳低位后翻及其他特征。有些患者伴尿道下裂和脊柱前移,但无晶状体异位。
- Thieffry-Kohler综合征表现为额部突出、小下颌、脊椎轻度侧凸、弓形足、趾重叠、跖骨囊肿。腔、踝骨质破坏起始于腕骨和跗骨并累及周围骨质,一般开始于儿童期并持续发展。骨溶解不对称,无疼痛。血清碱性磷酸酶和羟脯氨酸升高。
- Mirhosseini-Holmes-Walton综合征表现为小头,智力严重障碍,视网膜色素退变,白内障,关节伸展过度,细长指(趾)及脊柱轻度侧凸,为常染色体隐性遗传。晶状体异位还可发生于Weill-Marchesani综合征、Ehlers-Danlos综合征、高胱氨酸尿和成骨不全病例等。
- 常见的弹力纤维病还有类MFS、家族性主动脉瘤、MASS综合征、晶状体异位、Shprintzen-Goldberg综合征和先天性挛缩性蜘蛛指(趾)等,应注意鉴别。
早期发现MFS可行早期干预,发生心血管畸形宜早期手术治疗。骨骼病变无特殊治疗。
Prader-Willi综合征
Prader-Willi综合征以肥胖/性腺功能低下/隐睾症/智力迟钝和肌张力低下为特征。出生时体重低于正常,伴严重的张力降低和新生儿期喂养困难。男性有隐睾症和性腺功能减退,以后肌肉紧张性逐渐改善,体重增加速度加快。3岁时体重明显增加,伴异常面型,生长发育曲线低于正常,手、足过小。多数PWS患者存在GH缺乏,患者食欲亢进,进食增加,患者3~5岁时开始出现肥胖和轻度行为异常。在儿童中期和青春期,肥胖更加突出,并出现固执、脾气暴躁。
胎儿酒精综合征(fetal alcohol syndrome,FAS)、PWS、脆性X染色体综合征(FRAX)和结节性硬化症(tuberous sclerosis,TSC)均存在行为障碍。PWS应与其他后天出现的肥胖综合征进行鉴别。在新生儿期,鉴别诊断应包括引起严重的肌张力降低的疾病,如Zellweger综合征、21三体综合征、Werdnig-Hoffmann病、先天性肌紧张性营养不良、先天性肌病和胎儿运动不能综合征等。
综合性治疗有一定效果,首先纠正患者的行为心理异常,肥胖者节食,加强运动。GH治疗(每天≥1mg/m2)可促进生长发育,增加骨骼肌重量、减少脂肪贮量。
Parry-Romberg综合征
Parry-Romberg综合征以脂肪营养不良/蜘蛛状指/先心病/软骨发育不良/血管畸形和视网膜血管炎为特征。Parry-Romberg综合征亦称Romberg病、Parry-Romberg病、面偏侧萎缩症、进行性面部偏侧萎缩综合征(progressive facial atrophy syndrome)、进行性面部萎缩性神经病综合征、进行性板层发育不良综合征、先天性半侧萎缩症、先天性一侧萎缩症等。本征可发生于各种年龄,常见于20岁以前的女性。起病隐匿,进展缓慢;但6~24个月间发展较快;病变部位以左侧面部为多,也可波及同侧全身各处,甚至包括内脏(脑、肾上腺、卵巢、肾);萎缩涉及皮肤、毛发、皮脂腺、皮下脂肪、结缔组织、肌肉甚至骨,但以皮下脂肪为突出。面部多涉及三叉神经分布区,如眶下部、下颌部、颊部等。病变性质为硬皮样改变,部分患者可伴有局限性硬皮病。伴毛发异常、趾(指)甲发育障碍、Horner综合征、兔眼、A-R瞳孔、瞳孔散大、眼肌麻痹、眼睑下垂、虹膜异色等;一些患者有中枢神经症状,如偏头痛、局限性癫痫、舞蹈病、手足徐动症、三叉神经痛、脑内钙化灶;部分患者可伴有进行性脂肪营养不良、蜘蛛状指征、先天性心脏病、软骨发育不良、颅内血管畸形和视网膜血管炎等。
本综合征的诊断无困难,但要注意脑部的形态与功能检查以确定其与本综合征的关系。脑部CT和MRI表现通常为同侧脑实质的钙化、萎缩、软脑膜的强化、深部白质及皮层下白质的异常、皮质增厚等,该病脑内白质病变周围脑实质的线条状钙化为特征性改变。必要时可行血管造影检查。脑电图检查对一些不典型的癫痫有确诊价值。
伴疼痛过敏者可用交感神经阻滞法治疗,糖皮质激素有一定疗效。如考虑手术整容应做面部的MRI检查以确定最佳整容方案,萎缩的面部可用脂肪填充。
Aarskog综合征
Aarskog综合征引起身材矮小/面容丑陋/生殖器畸形。近年发现,Aarskog综合征的致病原因之一是FGD1基因突变。典型表现是:①身材矮小。②面容丑陋:前额突出,眼距增宽、低鼻梁、鼻孔前倾、耳厚而低位、上唇长而薄、人中发育不全、两上睑下垂。③隐睾、睾丸下降不全、阴囊褶包于阴茎的基部。④其他先天畸形(如心脏畸形、先天性远视等)。⑤有些患者的骨龄延迟,长骨干骺增宽,第5小指发育不良,第1掌骨和第1跖骨短而宽。⑥一般智商正常。Lebel等报道,FGD1突变(P312L)后的Aarskog综合征表现不一定很完全,患者可无颅面或生殖器畸形表现。本征应与22q11缺失综合征、Robinow综合征等鉴别。本征的预后欠佳,可存活至成年。虽早期生长发育迟缓,但青春期后有可能恢复正常发育。予以对症治疗,骨龄延迟及骨密度降低者可应用GH治疗以提高最终身高。
Sotos综合征
Sotos综合征伴过度生长/骨龄提前/智力落后/痴呆/特殊面容。Sotos综合征亦称巨脑症(cerebral gigantism),是一种异常的生长发育性疾病,表现为出生时体重过重,在生后1年内过度生长,骨龄提前、智力落后、痴呆和其他神经症状以及特殊面容,包括巨长头、双颞部毛发退化、眶距过宽及下颌突出,但最终身高、体重正常。
Sotos综合征与NSD1基因微缺失和突变有关。NSD1基因在生长和大脑发育中起重要作用。典型的Sotos综合征血清IGF-Ⅱ、IGFBP-3和IGFBP-4降低,而血清IGFBP-3水解增加。
伴有头围扩大的临床情况主要有脑水肿、神经纤维瘤病、软骨发育不良症、常染色体显性遗传性巨颅症等。虽然有些综合征也出现过度生长并伴有巨颅症,但Sotos综合征具有独特性,故鉴别多无困难。Nevo综合征除有类似Sotos综合征的特征外,还包括出生时全身水肿、严重肌张力减退、足挛缩、手腕下垂和指弯曲。Weaver综合征有独特的面部特征,长骨远端变宽和骨成熟加快,但两者的鉴别有时十分困难。Bannayan-Riley-Ruvalcaba综合征有Sotos综合征的一些特点,但有独特的阴茎色素斑和肠息肉(尤其是结肠)。临床上有些患者怀疑为Sotos综合征,而实为脆性X综合征。
Bloom综合征
Bloom综合征伴类红斑狼疮样皮疹/日光敏感/先天畸形及肿瘤倾向。Bloom综合征亦称面部红斑矮小综合征(facial telangiectasis-dwarfism syndrome)、蝴蝶状红斑综合征、染色体脆弱综合征、染色体断裂综合征、染色体不稳定综合征、先天性毛细血管扩张性红斑及生长矮小综合征、先天性毛细血管扩张性红斑综合征、侏儒面部毛细血管扩张综合征、侏儒-日光敏感-血管扩张性红斑综合征。为常染色体隐性遗传疾病,其特征是面部类似红斑狼疮样蝶形分布的毛细血管扩张性红斑,伴垂体性矮小症。
患者幼儿起病,类红斑狼疮样皮疹,日光敏感,矮小及其他先天畸形及肿瘤倾向。实验室可见IgA、IgM降低,IgG正常,淋巴细胞功能降低。染色体断裂率高,多呈四射体为本征特异表现。病变与BLM基因突变有关。一般先用免疫杂交和免疫组织化学方法筛查为阳性后再作BLM基因突变分析。
Bloom综合征患者的基因组不稳定。这些患者的B淋巴细胞株的微核(micronuclei)和姐妹染色体交换(sister chromatid exchanges,SCE)频率增高,对细胞毒药物有抵抗,易形成“花斑样转位嵌合体”(variegated translocation mosaicism)。(廖利珍)
