
确立矮小症的特征与类型
根据躯干和四肢的发育比例,一般分为匀称性矮小、肢短性矮小及躯干短缩性矮小3种类型,而判定的指标为下部量、上部量和臂间距。
首先测量儿童的实际身高(头顶至足底的长度)。若低于同年龄、同性别正常儿童身长的最低值,可视为身材矮小。同时要测量身体各部分比例和上部量与下部量。头顶至耻骨联合上缘为上部量,代表头颈部及脊椎部骨骼的生长状况(躯干长度);自耻骨联合上缘至足底为下部量,代表下肢长骨的生长状况(下肢长度)。12岁时,上、下部量的比例应为1∶1。身材矮小者的身长低于正常3个标准差,或在第3百分位数以下。如儿童身长增长<0.3cm/月,半年内<2cm,常为非体质性矮小所致。将身长增长速度动态观察绘制成曲线并与正常同龄儿比较,判断生长速度有无加快或落后。如为矮小症,应根据上部量与下部量的比例,确定其为匀称性矮小、短肢性矮小或躯干短缩性矮小。
匀称性矮小
以GH不足/缺乏、体质性矮小、非GH相关性矮小和体质性生长发育延迟多见,其次为各种重症慢性疾病及营养缺乏性疾病,而腺垂体疾病、女性(染色体性别,46,XX)性发育障碍、卵巢功能不全症等较少见,身材矮小的鉴别流程见图3-12-7。但需注意,男性(染色体性别,46,XY)性发育障碍的下部量明显超过上部量,而身高可正常甚至出现身材过高。
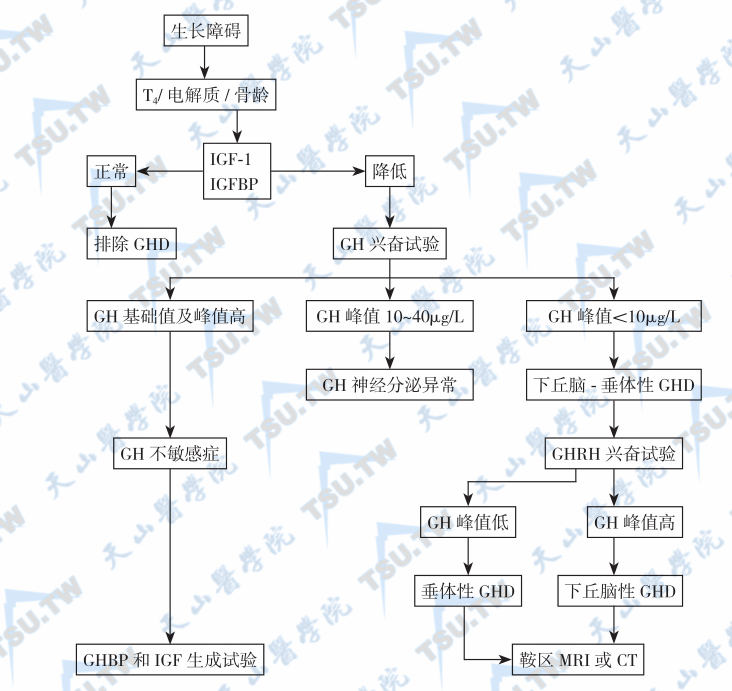
身材矮小的鉴别流程
肢短性矮小
一般与GH缺乏无关。下部量和臂间距明显缩短,下部量明显小于上部量,且不随年龄而增长,常提示为软骨不发育症(achondroplasia)、软骨发育低下症(hypochondroplasia)、致死性软骨发育不良症(thanatophoric dysplasia)、重症软骨不发育伴躯体发育延迟及黑棘皮病(severe achondroplasia with developmental delay and acanthosis nigricans,SADDAN)或颅缝早闭等。其次为SHOX基因突变、Leri-Weill综合征、海豹儿与儿童型甲减。如果患者的血清GH正常,且能排除甲减、海豹儿、Leri-Weill综合征,则需要对SHOX基因突变进行筛选和鉴定。在特发性身材矮小症中,2%~15%是由于SHOX缺乏症(SHOX deficiency)所致,后者亦称为软骨-骨生成障碍(dyschondrosteosis)。诊断的步骤是先用多倍连接依赖性探针扩增(multiplex ligationdependent probe amplification,MLPA)分析类型,然后做致病基因筛查。SHOX主要在胚胎和长骨生长板的肥厚性软骨细胞中表达。SHOX突变的外显率高,但临床表现很不一致,一般随着增龄,身材矮小越来越明显,女性较男性严重,生长迟滞于儿童开始,身高增长分析可发现身体的比例异常。GH的促生长作用类似于Turner综合征。
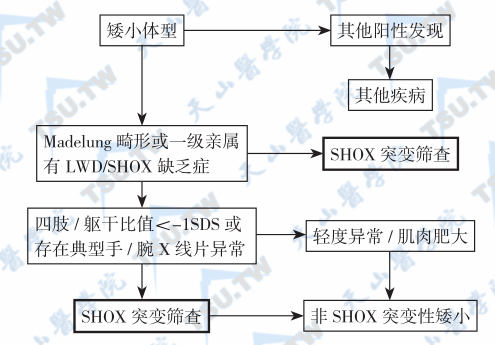
SHOX缺乏症的筛查流程
注:部分特发性身材矮小症是由于SHOX缺乏(SHOX deficiency)所致。诊断的步骤是先用MLPA 分析其类型,然后做致病基因筛查
躯干短缩性矮小
引起躯干短缩性矮小的主要原因为脊柱发育不全和黏多糖病,其次为多发性硫酸酯酶缺陷症、全身性神经节脂苷沉积症、甘露糖苷增多症、岩藻糖病、天门冬酰氨葡萄糖尿症,偶见于黏脂病、Kneist综合征及POEMS综合征等。
排除非GH相关性矮小症并鉴别其病因
在临床上,本病需与非GH相关性矮小症相鉴别。非GH相关性矮小症共同临床特点是:①对GH治疗无效(抵抗),但此种GH抵抗与前述的GH抵抗综合征不同,血GH和GH受体正常,发生GH抵抗的原因可能与控制生长发育的基因缺乏或失活有关。除儿童的自然生长外,GH无促进身高生长的附加效果,对IGF-1治疗亦无效或仅有微弱作用;除营养不良和全身性疾病所致的矮小症外,营养不良和运动治疗的效果也很差。②矮小症可伴由躯体畸形,但一般无肥胖,躯体呈均匀性细小(small figure)。③最终身高依赖于是否有青春期发育以及青春期发育的年龄。
慢性疾病
患者在儿童时期患有心、肝、肾、胃、肠等慢性疾病或各种慢性感染(如结核病、血吸虫病、钩虫病等),都可因生长发育障碍而致身材矮小。在儿童期和青春期,包括任何器官系统在内的严重慢性疾病均可引起生长不良。一些病例仅表现为生长延迟,而在另一些病例中,可通过改善营养来促进生长。患有胃肠疾病、肾脏疾病或癌症的患者,可通过胃肠外营养得到改善。引起生长发育障碍和身材矮小的其他疾病还有囊性纤维化、慢性胰腺炎、肺功能不全、先天性心脏病、心肌炎、充血性心衰、腹腔疾病、慢性血液病、幼年类风湿、慢性肾病、低磷血症、肾小管性酸中毒、神经性厌食等。
对身材矮小而其他方面正常的患者在行内分泌检查之前,应对其血红蛋白、白细胞计数、红细胞沉降率、血清胡萝卜素和叶酸水平、血清抗麦胶蛋白抗体或抗肌内膜(antiendomysial)抗体、血浆碳酸氢盐水平以及肝、肾功能进行检查。
Turner综合征与Noonan综合征
Turner综合征病因为X染色体缺如或畸变,患儿有性幼稚和身材矮小。除身材矮小外,伴有生殖器官发育不全和原发性闭经,亦可伴有颈蹼、肘外翻、盾形胸等畸形。血清GH正常。典型的45,XO性腺发育不全的Turner综合征正确诊断并不困难,而任何表型伴身材矮小女性都可能是Turner综合征的变异型。因此,如果没有发现导致体型矮小的其他原因,应行染色体核型检查。Noonan综合征(假性Turner综合征,部分患者的病因为PTPN11基因突变)有许多Turner综合征的表现特征,但女性核型为46,XX,男性为46,XY伴Noonan综合征,其他特征明显不同于Turner综合征。
早产儿和宫内生长迟缓
病因很多,如染色体病变、药物、先天性巨细胞病毒感染、胎盘缺陷等,亦可能与IGF的分泌及作用失调及IGF/IGFBP功能异常有关。与孕周成比例的宫内生长迟缓(IUGR)儿常伴随终生的身材矮小。1~2岁前,相应孕期的早产儿通常能赶上正常儿的身高和体重。但早产婴儿及出生时体重<800g者(孕期相一致),至少在第3年后,可能仍生长延迟,但骨龄、青春期开始的年龄、年生长速度正常,患者体形瘦小,可伴有Russell-Silver综合征。出生时为小样儿,三角形脸,肢体末端不对称,第5指(趾)弯曲等。巨细胞病毒、风疹病毒、弓形虫和人类免疫缺陷病毒引起的宫内感染可导致子宫内生长迟缓。母亲若使用药物,例如可卡因、苯妥英钠或怀孕期饮酒,均可引起IUGR。
常染色体基因组型异常及其综合征
许多常染色体基因组型的异常和伴或不伴智力发育迟缓的儿童畸形综合征(如Down综合征),身材矮小、体型不对称,四肢矮小,上部量和下部量的比例失调及上肢指距与身高不一致等。
骨发育异常
已知有100多种遗传性骨骼发育异常(骨软骨发育不良)。部分出生时即有肢体和躯干短小。常染色体显性遗传性软骨发育不全表现为四肢短小,头大并伴随有前额突出、鼻梁凹陷及腰椎前突。表现从严重的肢体矮小到外表基本正常不等,但青春期后生长减慢,导致成人身材矮小。
心理社会性矮小
心理社会性矮小(psychosocial dwarfism)一般有下列特点:①患儿遗传的体格生长和精神发育的能力正常;②环境因素使精神、心理、情感受到创伤,患儿所受的心理社会应激影响大脑皮质向下丘脑的神经冲动传递,抑制GHRH的分泌;③改变环境因素后可获得恢复。
早老症身材矮小
早老症(progeria,senile nanism)的病因不明,可能与中胚叶发育不全或内分泌代谢障碍有关。有家族遗传倾向,其临床特点为矮小症、消瘦、面似老人。患者头大,鼻尖突出,耳廓大,下颌骨较小。出牙较迟,颈部短而细,胸较狭,手足较小,但身体各部发育仍对称。长骨端骺部闭合较早,皮肤疏松多皱及色素沉着,皮下脂肪较少。性器官发育不全,第二性征和性欲缺如。也可有眼底与视神经病变,但智力正常。因全身性动脉粥样硬化,可有发作心绞痛,甚至心肌梗死,也可发生肾动脉硬化,肾功能不全或脑血管意外。
脑中线-视神经发育不良症
脑中线-视神经发育不良症(septo-optic dysplasia,SOD)属于罕见的先天性发育异常性疾病。临床上以脑中线发育异常(midline abnormalities in central nervous system)、视神经发育不良(optic nerve hypoplasia)和下丘脑-垂体发育障碍三联征为特征。病因与同源框基因(homeobox gene)Hesx1和SOX2突变有关,伴有眼、前神经板(anterior neural plate)和前脑发育不良。中枢神经中线异常主要包括胼胝体不发育、小脑发育不全、脑裂、透明隔缺如等。MRI可见脑组织形态异常、部分脑结构和神经垂体异位,严重者可能存在癫痫、偏瘫、智力障碍等。垂体激素缺乏和矮小较常见。因存在多种发育缺陷,虽然GH缺乏,但GH补充治疗的效果不佳。
特发性矮小症和体质性矮小症
特发性矮小症(idiopathic short stature)和体质性矮小症(constitutional short stature)是指个体的身高低于同种族、同地区、同年龄、同性别人群平均身高的2个标准差,而矮小的原因未明者。特发性矮小症可进一步分为家族性和非家族性两类。其临床特点与GH缺乏性矮小症基本相同。家族中有多名成员矮小(部分为散发性),但无器质性疾病,骨龄正常;无系统疾病。GH分泌正常,GH兴奋试验可显示反应低下。患者对GH治疗有一定反应。特发性矮小症的诊断必须在排除畸形、骨发育不良症、袖珍儿(低体重儿)、内分泌性疾病和系统性疾病等所致身材矮小后,方可成立。
体质性矮小症的诊断标准是:①无系统疾病,营养正常;②体格检查无异常发现,躯体的比例正常;③血TSH、T3、T4 和GH正常;④身高在第3个百分位数内,但年生长率在第5个百分位数以上;⑤青春期发育延迟;⑥骨龄延迟;⑦成年后可达到预期身高。
根据病史和特殊检查鉴别GH相关性矮小症病因
GH相关性矮小症的病因很多(表3-12-5)。遗传性GHD可有家族史,呈常染色体隐性或显性遗传,并能查出相应的突变基因。GH抵抗综合征的病因可能来源于GH、GH受体或GH受体后,患者血GH很高且有活性,血IGF-1降低,外源GH无促生长作用,GH的生成、分泌或生物活性异常。特发性GHD有围生期病变,用GHRH兴奋试验可鉴别IGHD患者颅内损伤的部位。继发性GHD主要有3种临床类型,其病因各异,GHD仅为腺垂体功能减退的表现之一,可由下丘脑-垂体区获得性病变如头颅创伤、肿瘤、感染、浸润性病变引起。
慎重鉴别GHD与特发性青春期发育延迟
特发性青春期延迟者往往有家族史,出生时体重正常,出牙稍晚,身材较同龄正常人矮小,骨发育可推迟数年,常到16~17岁以后才开始第二性征发育。智力正常,无内分泌系统或慢性疾病依据。一旦开始发育,骨骼生长迅速,性成熟良好,最终身高可达正常人标准。对青春期发育显著推迟,尤其是伴有增长明显延缓或停滞者,应做颅骨X线检查、眼底、视野以及系统的神经检查,口颊黏膜涂片检查性染色质和尿或血浆促性腺激素测定助于诊断。
