
英文:thrombocytopenia with absent radius syndrome (MIM 274000);
同义名:四肢短-血小板减少综合征,无桡骨血小板减少综合征,血少板减少伴桡骨发育不全综合征,Gross-Groh-Weippl综合征,TAR综合征。
溯源与发展
1956年由Gross、Groh、Weippl三人在奥地利首次报道的一组以桡骨异常为主,合并心脏等畸形的综合征而命名。1969年Hall对31个家系进行调查,发现以女性患者占多数。亦有的学者认为,只有血小板减少而无红细胞和白细胞减少,并伴有桡骨发育不全,是Fanconi综合征的一个亚型,是其多种遗传表型之一。
发病机制
确切病因和发病机制未明。一般认为是由基因异常所致的先天性组合畸形。
遗传学:遗传方式属AR,后代再显风险率为25%;女性患者比男性发病率高,其原因不明。致病基因定位于17q21.3。
临床表现
患者在新生儿期常有出血点和紫癜以及危及生命的出血发作。血小板减少是发作性的,且常为非特异性感染和紧张所诱发,故除大出血外无明显贫血。一般来说红细胞和白细胞正常,部分患者可经常出现类白血病反应且伴有肝脾肿大。几乎所有病例均可见双侧桡骨发育不良或缺如,少部分患者为单侧受累。亦可有其他骨骼畸形,如桡侧棒状手,掌指骨发育不良,偶有尺骨发育不良或缺如,肱骨异常,肱骨或脊柱畸形,髋错位和上下,颌骨发育不良,三角头等。双侧拇指发育正常。继发于颅内出血而致精神发育迟缓,牛奶过敏,出汗增加,部分患者出现腹泻。
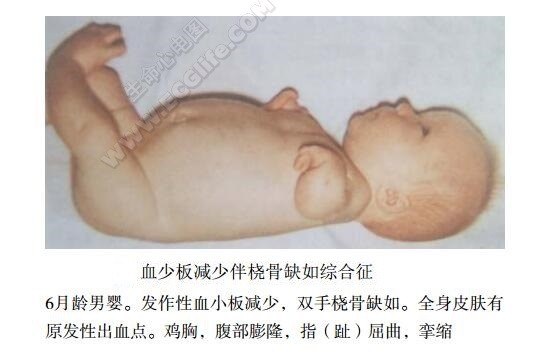
心血管损害:30%伴有心血管畸形,以房间隔缺损、法洛四联症、右位心为常见,患者可有心悸、气短、无力、心脏扩大和心杂音等表现。法洛四联症患者出现紫绀和杵状指。单纯右位心患者则多此症状,而仅有X线和心电图等改变。
诊断、鉴别
- 诊断:依据血小板严重减少(仅及常人的1/2~1/3)以及桡骨发育不全或缺如两大临床主征,结合骨髓检查显示巨核细胞减少或缺如,即可诊断。
- 鉴别:需与先天性全血细胞减少症相鉴别,本病仅有血小板减少,后者为全血象减少。本病骨骼异常以双侧桡骨发育不良为主,后者除前臂骨骼发育不全外伴有拇指发育不良。还必须与遗传性心血管-上肢畸形综合征鉴别,此病无血小板减少,骨骼损害以拇指系统为主,伴有前臂骨桡侧为重,可资鉴别。
治疗
产前:妊娠第20周超声波或X线检查能发现骨骼异常;生后:无特殊治痕随时监测血小板计数,当需要时可酌情输入血小板或新鲜血液,以纠正致命性出血。尽早做矫形手术,使手畸形和心血管异常获得矫正。
预后
主要取决于血小板减少的程度,其与出血症状呈正比,常是死亡的主要因素。不少患者在婴儿期死于突发性大出血,出生后第一年内死亡率高,随年龄增长死亡率降低,成人时血小板数可接近正常。产前诊断可预防病儿出生,在妊娠2个月内,X线和超声可证实上肢畸形,第3个月可通过取胎儿血测出血小板数,如少于50x104/mm2,严重出血的危险性增高,应考虑人工流产。
