
BMD升高和反复骨折伴贫血与软组织钙化及颅骨增厚是骨质硬化的重要诊断线索。在临床上,遇有下列情况者要考虑骨质硬化症的可能,并进行进一步检查明确诊断:①BMD升高;②反复骨折而BMD正常或升高;③不明原因的贫血、血小板减少或者拔牙后反复感染;④软组织钙化或颅骨增厚;⑤自发性单神经瘫痪。
本病的遗传特点为:父、母发病,遗传上无明显性别差异,其子女发病概率相仿。对于常染色体显性遗传骨硬化症,常见外显不全现象,确切外显率尚待明确。典型病例的诊断依据是:
- 贫血、出血、发育不良、抵抗力低和易并发感染;
- 查体发现肝、脾大,同时有视力、听力下降;
- X线表现为普遍的骨皮质增厚、骨小梁消失、BMD增高及骨髓腔变窄;
- 血清钙、磷、ALP,尿羟脯氨酸正常,在排除下列以骨质硬化为突出表现的其他骨病后诊断可以成立,但是中间型骨硬化患者可以呈现显著高的ALP;
- 少数诊断困难者可作MRI、CT、骨组织形态计量等检查;
- 病因诊断有赖于对候选致病基因进行序列鉴定分析。
骨质硬化伴贫血/脑钙化/肝脾大/病理性骨折/顽固性骨髓炎
骨质硬化的治疗困难,病情呈进行性发展。晚期病例多伴有贫血、脑钙化、肝脾大。骨质硬化的主要危险是病理性骨折和顽固性骨髓炎,最常见的是颌骨骨髓炎,治疗相当困难。骨质硬化增生的骨孔可压迫神经,导致瘫痪、耳聋或视力下降。
根据X线片和BMD测量确立骨质硬化症诊断
严重病例的诊断较易,一般凭X线照片即可诊断。有困难时可作CT或MRI检查。其X线表现较具特征性,虽然不同亚型的X线有一定差异性,但大部分表现相同:①弥漫性骨质硬化可见于所有骨骼。如为均一性,可使长骨呈“棒球棒”样或粉笔样改变;如表现为硬化带与透明带交替排列,在髂骨翼及邻近的长骨近端呈“年轮状”,在脊椎表现为“夹心面包样”或“骨中骨”症;在其他骨均表现为“骨中骨”症。②颅骨增厚,致密,以颅底部较明显,鼻窦小而气化差。③可伴骨折和骨髓炎。
血钠、钾、钙、磷、镁多正常,血清酸性磷酸酶一般升高。儿童中有低磷酸盐血症并偶尔可见中度的低钙血症。成人血钙和碱性磷酸酶值则一般正常。此外,在婴幼儿恶性型骨质硬化症有血清肌酸激酶BB升高,血清OPG和RANKL的比例升高,血清C末端肽和尿N末端肽的浓度降低。Ⅱ型成年型骨质硬化症患者中可见血清抗酒石酸酸性磷酸酶(TRAP)升高。
轻型病例的X线照片和CT正常,仅见BMD升高。重型患者的整个骨骼系统呈硬化表现,这类患者常显示血ALP显著增高。轻型病例往往在BMD测量中被发现,遇有不明原因的BMD升高者都应想到骨质硬化症可能。结合临床表现,进行致病基因鉴定和突变分析可明确病因诊断。
单纯型骨质硬化症与复合型骨质硬化症的鉴别
单纯型骨质硬化症已如前述。复合型骨质硬化症所伴发的疾病包括幼白成红细胞增多症、血小板减少、肾小管酸中毒、外胚层发育不良与免疫缺损、白血病黏附缺陷综合征、肢骨纹状肥大症、先天性条纹状骨病以及矮小症等。
复合型骨质硬化症分类

注:其他致病基因可能包括PLEKHM1、IKBKG、NEMO/Kindlin-3、CalDAG-GEF1、LEMD3、WTX等。除迟发型骨质硬化(Albers-Schönberg病)的基因突变机制属于优势负性(dominant negative)抑制外,其他均为失活性突变(loss of function)。Pleckstrin:血小板白血病C激酶底物。
Engelmann-Camurati病
Engelmann-Camurati病(Engelmann-Camurati disease,ECD)呈常染色体显性遗传,早年发病,以长骨对称性进行性骨干发育不良症(progressive diaphyseal dysplasia)和矮小为特征。病因与TGF1基因突变有关。50%以上的CED患者可单独用X线照片获得诊断,表现如股骨增宽,密度明显增高,偶尔伴有轻度的颅骨损伤,但无其他骨畸形,诊断有困难时首用CT证实为骨干病变,并了解脑神经是否受损伤。
Camurati-Engelman病需与下列疾病鉴别:
- Lenz-Majewski骨发育不良(Lenz-Majewski dysplasia);
- Kenny-Caffey骨发育不良(Kenny-Caffey dysplasia);
- 青少年型Paget骨病(Juvenile Paget’s disease of bone);
- Van Buchem骨发育不良(Van Buchem dysplasia);
- 多发性(multiple epiphysal dystrophy,Ribbing病)。
多发性骨干硬化症
多发性骨干硬化症(multiple diaphyseal sclerosis)是Ribbing于1949年描述的非对称性骨干硬化性病变,亦称Ribbing病(Ribbing disease,RD),是一种少见的良性骨发育不良症,目前共有20多例报道。特点是骨内膜新骨形成,骨硬化仅局限于下肢长骨骨干,多见于青春期后年轻人。病因未明,除不发生神经系统损害外,其他表现与Camurati-Engelmann病相似。起病之初主要表现为骨痛、常误诊为骨髓炎或骨形成骨肿瘤(bone-forming neoplasia),需要鉴别的疾病有骨肉瘤、骨样骨瘤(osteoid osteoma)、骨髓炎和应激性骨折等。诊断困难时,可用MRI或99mTc-MDP骨扫描显示骨损害,明确诊断;症状明显者可以非甾体抗炎药或二膦酸盐治疗。
多发性干骺-骨骺点状硬化症
多发性干骺-骨骺点状硬化症(osteopoikilosis)是一种常染色体显性遗传的良性的干骺-骨骺硬化症,少数病例散发。病因未明,但与软骨内成骨异常有关。多数多发性干骺-骨骺点状硬化症病例仅在意外中发现,硬化灶1~2mm,最大可达10mm;硬化灶位于关节周围的干骺端硬化灶科可形成骨岛或内生骨疣。如果患者没有症状,不需要特殊处理。但当患者存在关节痛、骨痛等症状时,应使用骨扫描排除其他病变尤其是前列腺癌骨转移可能。除多发性干骺-骨骺点状硬化症外,能引起软骨内成骨发育不良的其他良性骨硬化症有内生骨疣(骨岛)早期骨质硬化症、致密性成骨不全症、条纹状骨病及条纹状骨病伴颅骨硬化症等,应注意鉴别。
软骨内成骨发育不良的鉴别

注:AD:autosomal dominant,常染色体显性遗传;AR:autosomal recessive,常染色体隐性遗传
在组织学和影像学上,内生骨疣(enostosis)与多发性干骺-骨骺点状硬化症极为相似,骨岛可能小或单独出现,小梁骨属于编织骨,骨质硬化与致密性成骨不全症或条纹状骨病易于鉴别。
硬化性骨狭窄与van Buchem病
近年,在硬化性骨狭窄(sclerosteosis,OMIM 269500)和van Buchem病(OMIM 239100)研究中发现了新的骨形成调节信号通路——骨硬化素(sclerostin)通路。在胚胎期,许多组织均可表达SOST mRNA,但只有矿化组织中的终末期骨细胞和肥大型软骨细胞才能表达骨硬化素,听囊(otic vesicle)和肢芽(limb buds)等非矿化组织的SOST mRNA表达异常可能与并指畸形有关,硬化性骨狭窄和van Buchem病不发生心肌、主动脉、肝脏、肾脏血管钙化,这些组织表达SOST mRNA的意义未明。骨细胞分泌的骨硬化素抑制成骨细胞骨形成。硬化性骨狭窄和van Buchem病首次报道于1950年,属于两种不同的硬化性骨病,但临床表现相似。主要见于非洲(南非)籍的荷兰人后裔,而van Buchem病多发生于荷兰的小渔村(small fishing village),但其他各地(西班牙、巴西、美国、德国、日本、瑞士、塞内加尔等)亦有零星报道。
在骨重建中,新形成的骨细胞分泌骨硬化素,转运到骨表面,抑制成骨细胞骨形成,避免骨重建单位多度矿化;Wnt信号与Wnts、卷毛受体、LRP5/6辅助受体形成复合物,β-连环蛋白增多,Dkk1拮抗剂通过LRP5/6和Kremen形成的复合物抑制Wnt信号,移除细胞膜上的LRP5/6。Dkk1与LRP5/6的第一和第三螺旋琨结合,而骨硬化素与LRP5/6的第一螺旋琨结合,抑制Wnt信号。
硬化性骨狭窄与SOST基因(17q12-21,编码骨硬化素,目前已报道了5种突变类型),而van Buchem病未发现与SOST基因突变相关。骨骼病变特点是骨内膜骨质增生(endosteal hyperostosis)并发展为进行性泛发性骨质硬化症(progressive generalized osteosclerosis),其中以颌骨、颅骨为主要侵犯部位,导致下颌和面部畸形、颅内压升高、脑神经受压。表现为听力下降、面瘫、嗅觉减退。硬化性骨狭窄的病情较重,常伴有并指畸形;骨活检发现,患者的骨形成增强,成骨细胞呈立方形,四环素标记的类骨质间距扩大,矿化正常。相反,破骨细胞活性和数目没能相应增加,从而发生骨质硬化。血清骨形成指标升高或正常。
高骨量综合征
LRP5基因含23个外显子,相隔>100kb,有至少13种序列多态性,但对其功能可能没有明显影响。LRP5活化性突变(G171V)患者伴有骨密度升高,称为高骨量综合征(high bone-mass syndrome)。文献报道的LRP5突变家系或病例见下表,LRP5基因编码区多态性与突变分别见下表。
文献报道的LRP5突变家系或病例


注:骨内膜骨肥厚:endosteal hyperostosis
LRP5基因编码区多态性
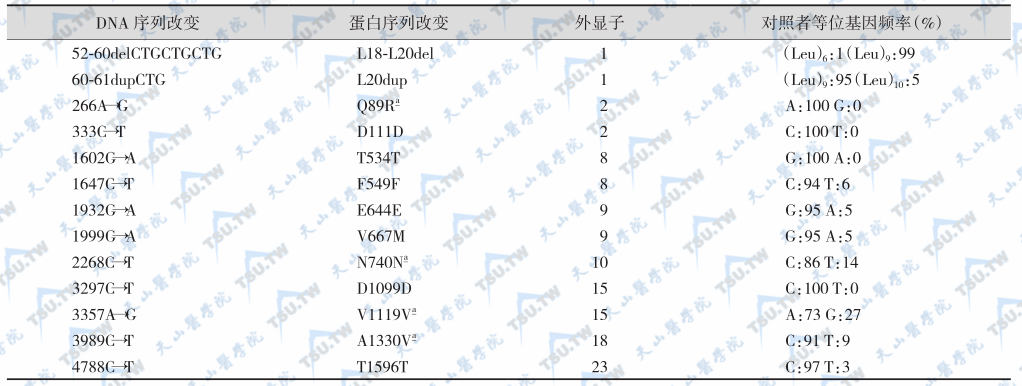
LRP5突变
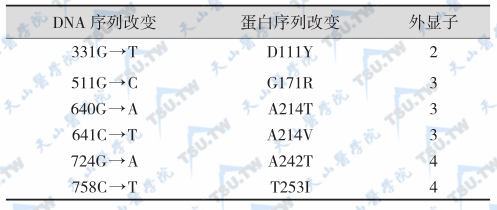
X线照片显示骨密度显著增加,常见的累及部位有颅骨、脊柱、骨盆和四肢长骨,严重病例的骨髓腔狭窄。硬腭骨隆凸和牙齿变化是高骨量综合征的重要特征,另一个特点是虽然骨密度明显升高,但极少发生骨折,这是与其他骨质硬化的显著差别之一,是具有重要鉴别意义。
肢骨纹状肥大症
肢骨纹状肥大症(osteopathia striata)是调节骨形成的Wnt途径因子突变所致。Ghosal综合征与厚皮性骨膜病(pachydermoperiostosis)是由于类二十烷酸途径(eicosanoid pathway)功能障碍引起的。肢骨纹状肥大症伴颅骨硬化(osteopathia striata with cranial sclerosis)是一种X性连锁遗传性骨病。表现为骨盆和长骨的干骺端条纹状骨硬化,往往伴有颅骨硬化。WTX基因(FAM123B)突变可能是其致病原因,WTX编码Wnt的抑制因子。患者常伴有额部前突、巨头、眼距过宽、鼻梁宽大、腭裂、听力减退和智力障碍。男性患者的病情重,常为致命性的;问题是WTX基因突变与许多肿瘤发病相关,但肢骨纹状肥大症的肿瘤风险并未增高。
Ghosal综合征
Ghosal综合征(Ghosal syndrome)亦称Ghosal血液-骨干发育不全综合征(Ghosal hemato diaphyseal dysplasia syndrome,GHDD)。患者骨密度升高,部分存在TBXAS1突变,TBXAS1基因编码的凝血 烷合酶(thromboxane synthase,TXAS)是一种花生四烯酸信号因子,可产生血栓素A2(thromboxane A2,TXA2)。患者的血小板花生四烯酸凝血过程障碍,并影响编码RANKL/OPG的TNFSF11 和TNFRSF11B基因表达。因而导致骨质硬化。
烷合酶(thromboxane synthase,TXAS)是一种花生四烯酸信号因子,可产生血栓素A2(thromboxane A2,TXA2)。患者的血小板花生四烯酸凝血过程障碍,并影响编码RANKL/OPG的TNFSF11 和TNFRSF11B基因表达。因而导致骨质硬化。
致密性成骨不全症
组织蛋白酶K(cathepsin K,CTSK)属于木瓜蛋白酶样-半胱氨酸蛋白酶(papain-like cysteine protease)家族成员,CTSK蛋白由前原区(15个氨基酸残基)、前区(氨基酸残基)和成熟肽(215个氨基酸残基)组成。突变的类型包括错义突变、框架移动、无义突变、剪接突变、终止密码子突变。CTSK基因突变引起常染色体隐性遗传性致密性成骨不全症(OMIM 265800)。分析文献报道的159例致密性成骨不全症(pycnodysostosis)患者的临床资料,共涉及59个家族33种突变类型;37.29%来源于欧洲,30.51%来源于亚洲。69.70%的突变发生于CTSK的“成熟结构域”,24.24%位于“前区”,6.06%位于“前前区”,突变热点在第6号外显子。CTSK突变使骨基质蛋白(如Ⅰ型胶原)的降解和骨骼形态异常。临床表现为身材矮小、骨密度升高、骨折、囟门未闭。
文献报道的致密性成骨不全症CTSK基因突变

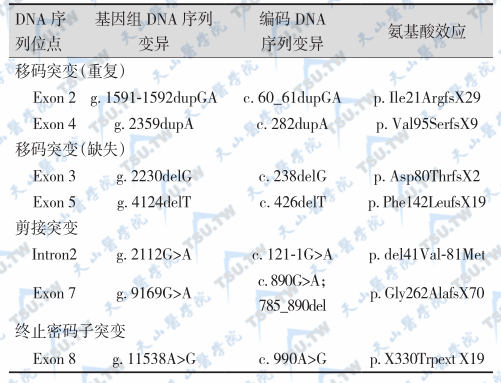
致密性成骨不全症的临床特征
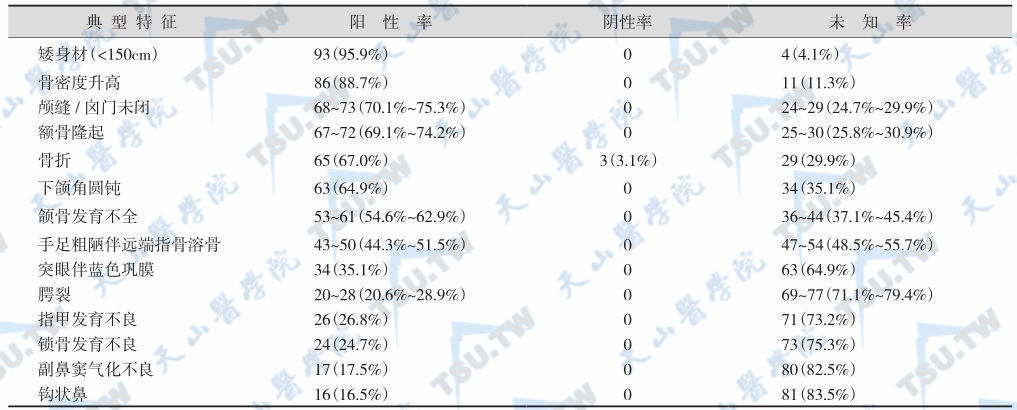
注:病例97例(男性42例,女性44例,未报道性别11例)
厚皮性骨膜病和肥大性骨关节病
厚皮性骨膜病(pachydermoperiostosis)是一种遗传性皮肤病,多于青春期发病。表型为进行性皮肤增厚、头皮松弛(cutis verticis gyrata)、杵状指、关节周围肿胀和长骨骨膜赘生。X线照片显示骨膜赘生、分叶状骨膜反应;膝关节亦可见骨膜赘生、骨皮质增生、关节腔渗液、股四头肌和膝盖骨钙化。病变发展缓慢(10~20年)。皮肤的其他表型有皮肤脂溢(seborrhea)、皮肤红斑、眼睑脱垂、痤疮、多毛。有些患者有关节痛、关节炎、关节积水、关节积血。X线照片显示有关节腔狭窄、软组织肿胀和指骨骨溶解。组织检查可见皮肤硬化、血管淋巴细胞侵润。临床上可分为三种类型:
- 完全型:以面部皮肤增厚和头皮松弛突出,伴有杵状指和原发性肥厚性骨关节病(primary hypertrophic osteoarthropathy);
- 不完全型:无皮肤增厚和头皮松弛;
- 顿挫型(fruste form):皮肤一处或多处改变,骨损害轻微或缺乏。
肥大性骨关节病(hypertrophic osteoarthropathy)主要见于发绀型先心病患者,始发于掌骨、跖骨和上、下肢长骨的远端。组织学的早期变化是骨膜、滑膜、关节囊与毗邻皮下组织水肿及圆细胞(round cells)浸润,新骨形成并伴有骨吸收加速;有时上、下肢长骨可出现酸痛、压痛或剧痛。在内分泌代谢病领域里,引起杵状指的疾病主要见于神经内分泌肿瘤(如GH瘤、类癌综合征等)。
杵状指(趾)与肥大性骨关节病的发病机制基本相同,因为两者的锝(technetium,Tc)代谢示踪观察与杵状指及长骨骨膜的亲和性研究结果与病理表现完全一致。发绀型先心病患者通过右向左分流,使巨核细胞胞质中的血小板生长因子(PVGF)及转化生长因子-β(TGF-β)作用于指(趾)的骨膜毛细血管。血小板生长因子是一种强效的有丝分裂原,能与反应细胞的受体结合,但因半衰期极短,所以仅发挥局部作用。这些细胞因子作用于骨髓间质细胞,促进蛋白合成,导致结缔组织增生及细胞增殖;而中性粒细胞、T淋巴细胞、单核细胞及成纤维细胞的趋化因子促进细胞外基质增生。
单纯型骨质硬化症与继发性骨质硬化症的鉴别
继发性骨质硬化症主要见于代谢性疾病或综合征,如Ⅰ型Gaucher病、B型Niemann-Pick病、膜型脂肪营养不良症、Ⅰ型耳-腭-指综合征等。
引起骨密度升高的原发性疾病
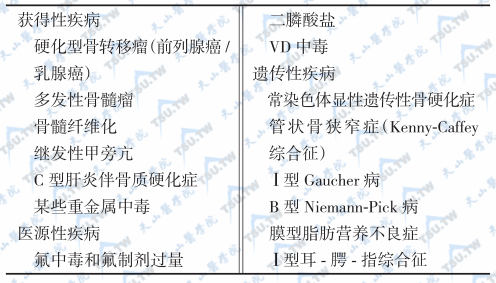
全身性骨质硬化症与局限性骨质硬化鉴别
由于调节骨代谢因子和成骨细胞与破骨细胞活性的基因突变引起的骨质硬化发生于所有骨骼,因而表现为全身性骨骼硬化,骨密度显著升高,骨脆性增加,极易发生骨折。局限性骨质硬化症的病因主要是骨发育不良所致,但亦涉及调节骨代谢的某些基因或细胞因子。
肢骨纹状肥大症
肢骨纹状肥大症(melorheostosis)是过早骨化和骨密度增高综合征中的一种,亦称Leri-Joanny综合征、Leri病、Leri流液状骨膜炎、Leri型脆性骨质硬化、蜡油样骨质增生症、流动性骨硬化症、流波状骨质硬化症、局限性骨密度症、蜡油状骨病、烛泪样骨质增生症、肢骨纹状肥大症、烛泪状骨髓炎、肢骨纹状增生症等。
肢骨纹状肥大症和骨斑症可合并存在于同一患者中,此时可诊断为混合性骨增生不良性硬化症(mixed sclerosing bone dysplasia)。一般认为,肢骨纹状肥大症为散发性发病,而骨斑症为常染色体显性遗传性骨病。Nevin等报道,1例肢骨纹状肥大症患者的母亲和妹妹有骨斑症,但无肢骨纹状肥大症,提示两种疾病可能有遗传上的某种联系。病变部位皮肤成纤维细胞的TGF-β诱导性betaig-h3、骨连蛋白、纤连蛋白、Ⅰ型胶原表达均被下调。这些黏附蛋白的表达异常可能是骨质硬化和软组织病变的病因之一。本症常幼年起病,进展快;但成年后发展缓慢。多数伴有先天性发育畸形。有人认为是常染色体显性遗传。骨、关节、软组织病变具有偏侧发病倾向。
骨外膜-骨内膜骨质增生,骨板排列密集紊乱,骨髓腔被纤维组织所代替。骨病变的特点是类骨质丰富,血管生成增多,类骨质矿化不良,骨吸收增强,破骨细胞数目增多。血管周围的骨膜成纤维细胞、间充质细胞、内皮细胞和成骨细胞可见较丰富的TGF-β表达,内皮细胞和肥大细胞还表达碱性FGF(bFGF)。
本征于1922年首先由Leri和Joanny报道。本征可发生于任何年龄,累及任何骨骼部位,但以四肢骨为多见。男、女发病率相当,起病隐匿,常于数年后出现骨骼畸形、疼痛、肢体僵硬、活动受限而就诊。Freyschmidt总结23例患者的临床资料,单一骨受累者10/23,2个骨骼受累者4/23,3个骨骼受累者9/23;上肢5/22,下肢16/23,上下肢均受累者1/23。放射学上有骨病改变者6/23,混合型病变者4/23。其临床特征是:①常见于5~20岁;②缓慢起病;③夜间休息、过劳、压迫后疼痛为早期表现;④病变呈萎缩纤维化,跖腱膜均明显增厚和广泛纤维化,伴严重萎缩和畸形,皮下组织因压迫性缺血可发生压疮;⑤可有神经合并症,如腕管综合征、关节疼痛、肿胀、活动受限、变形;⑥肢体缩短较常见,使骨纵向生长缓慢。骨病变局部皮肤张力高,呈红斑样改变,累及的肢体挛缩、变短。有些病人可合并条状硬皮病(linear scleroderma)。严重的肢骨纹状肥大症患者可侵犯病变同侧肢体,肢骨纹状肥大症因硬化性骨发育不良,X线片上可见条纹状骨肥大,犹如蜡烛滴样,一般位于长骨的一侧,并可引起骨痛、活动障碍和肢体不对称。
全身性脆性骨质硬化
是一种少见的常染色体显性遗传病,其特点是长骨、腕骨、跗骨等处骨干和干骨骺端出现多发性脆性骨质硬化斑点。LEMD3基因杂合子突变是本病的主要病因。Buschke-Ollendorff综合征(BOS)的特点是无症状的全身性脆性骨质硬化伴皮肤结缔组织痣(connective tissue nevi)。此外,全身性脆性骨质硬化可与局限性吝状骨过多症(melorheostosis,MRO)合并存在,使患者的骨病损害更严重,但不是所有的局限性吝状骨过多症患者都有LEMD3突变。Sevjidmaa等发现,在同一家族中,LEMD3与EXT1突变可分别导致全身性脆性骨质硬化(osteopoikilosis)和多发性外生骨疣。
常单侧长骨皮质增生,亦可见于肩胛、骨盆、脊柱、肋骨、头颅(少见)。沿长骨骨干线条状骨皮质增生,类似向下流注的蜡油。增生可波及髓腔,软组织内小的骨质沉着(特别是关节),脊髓生长线早期闭合。增生性病变可导致局部动脉受压或闭塞。
本病的病变主要在四肢的长管状骨,外膜增生明显,类似流注的蜡油,可具有疼痛、关节僵硬、四肢不对称、肌肉消瘦等症状。如诊断有困难,可作CT或MRI检查以确定骨与软组织的病变关系。本征应与其他骨质硬化性疾病相鉴别,尤其应与全身性脆性骨质硬化(osteopoikilosis)、条纹状骨病(osteopathic striata)、骨化性肌炎(myositis ossificans)、骨旁骨肉瘤(parosteal osteosarcoma)或骨瘤(osteoma)等鉴别。
颅缝早闭综合征
颅缝早闭(craniosynostosis)是一组散发性常染色体显性遗传的异质性疾病,具有明显的临床重叠表现。细胞的生长、分化、移行、血管的生成、伤口的愈合和肿瘤形成都离不开成纤维细胞生长因子(FGF)的调节。在细胞水平上,FGF通过FGFR的介导而发挥作用。FGFR有四种亚型,分别由不同的基因编码,即FGFR1~4。目前已发现FGFR1~3三个基因在人类均存在基因突变而引起的疾病,已多达数百种,其中主要有Apert综合征、Beare-Stevenson综合征、Crouzon综合征、Jackson-Weiss综合征、Muenke综合征、Pfeiffer综合征和Saethre-Chotzen综合征。这些综合征的共同特点是颅缝早闭,其共同的分子病因是FGFR1~3基因的突变(Saethre-Chotzen综合征还可由FGFR的相关基因——ACS基因突变引起)。另一方面,从颅缝早闭的病因上看,除上述的遗传性疾病外,还可由许多非遗传性疾病引起。例如,FREM1基因的拷贝数变异(CNV)与突变亦可引起颅缝早闭。
额骨提前融合引起额缝早闭(metopic craniosynostosis,MC),颅骨三角畸形。病因可能与FREM1基因的拷贝数变异(CNV)或突变有关。
Pfeiffer综合征
1964年,Pfeiffer描述的综合征包括颅缝早闭、宽拇指、宽 趾和各种其他特征及手部软组织并指,三代中有8个亲属受累。此综合征遗传类型为染色体显性,外显率高,表型各异。病因主要与FGFR2和FGFR1基因突变有关。
趾和各种其他特征及手部软组织并指,三代中有8个亲属受累。此综合征遗传类型为染色体显性,外显率高,表型各异。病因主要与FGFR2和FGFR1基因突变有关。
头颅通常为尖短畸形,在某些病例中有颅面不对称、上颌骨发育不足和相对下颌前突,鼻梁凹陷,眶间距过宽,睑裂下斜,眼球突出,外斜视等。颅骨三维重建显示双侧冠状缝和左侧人字缝硬化,面骨发育不良、鼻呈钩状型、腭盖高拱、牙槽突扁平、牙齿拥挤。某些受累患者颅面不对称,静脉显露。拇指和 趾宽大,并常伴有足外翻畸形,软组织并指(趾)畸形,常累及手和足的第2、3指(趾),有时累及3、4指(趾),但短指(趾)可在无并指(趾)畸形的病例中见到。偶见指(趾)弯曲畸形。手和足常可见指(趾)短小,有些病例可见中指(趾)缺乏,
趾宽大,并常伴有足外翻畸形,软组织并指(趾)畸形,常累及手和足的第2、3指(趾),有时累及3、4指(趾),但短指(趾)可在无并指(趾)畸形的病例中见到。偶见指(趾)弯曲畸形。手和足常可见指(趾)短小,有些病例可见中指(趾)缺乏, 趾的远中趾骨宽,近中趾骨畸形,第一跖骨变宽、短小,在一些病例中可见双跖骨。
趾的远中趾骨宽,近中趾骨畸形,第一跖骨变宽、短小,在一些病例中可见双跖骨。
在X线照片上,脊柱MRI显示尾骨反转,脊柱的生理弯曲消失。第1、2跖骨的副骺和指趾的近中趾骨有两个钙化中心。 趾偶见部分双趾。一些病例可累及掌骨和跖骨近端的腕骨和跗骨融合。
趾偶见部分双趾。一些病例可累及掌骨和跖骨近端的腕骨和跗骨融合。
普通型Pfeiffer综合征患者的智力一般正常,但在一些病例中可见智力缺陷。脑水肿、Arnold-Chiari畸形、癫痫发作亦可见到。此综合征常伴有苜蓿叶状头颅畸形,并出现其他在Pfeiffer综合征普通型中不易见到的各种畸形。常可在婴儿期死亡。一些患者可见颈椎和腰椎融合。肱骨短、肘外翻、桡骨肱骨和桡骨尺骨融合、骨盆畸形、髋关节外翻和马蹄形内翻足也偶有报告。少见的异常还包括有幽门狭窄、脐疝、肛门阴囊基部异位、阴囊分叉、乳头间距宽、眼睑下垂、瞳孔异位、角膜瘢痕、视神经萎缩、漏斗闭锁、耳前赘生物(结节)、外耳道缺如、听力缺陷、悬雍垂裂、多生牙和牙龈肥大等,根据典型表现一般可作出临床诊断,但分子病因有赖于FGFR基因分析。
Crouzon综合征
Crouzon综合征(OMIM #123500)由Crouzon于1912年首先描述,其特征性的临床表现有颅缝早闭、上颌骨发育不全、眼眶浅和眼球突出。Crouzon综合征为常染色体显性遗传,67%的病例有家族倾向,33%散发,发病率约1/25 000。Crouzon综合征的表现具有多样性特征。先证患者可仅表现为苜蓿叶状颅骨(家族成员中受累最重者),但后代患者表现有典型的Crouzon综合征。受累的母亲和家族中的其他成员表现有眼球突出和面中部缺陷,无颅缝早闭。现已查明,本综合征属于遗传性颅缝早闭中的一种,其发病主要与FGFR2基因突变有关。Crouzon综合征的临床表现见下表。
Crouzon综合征的临床表现

Crouzon综合征颅骨畸形取决于颅缝闭合的顺序和速度。尖颅是最常见的表现。也可见狭颅、三角形颅及苜蓿叶状颅。颅缝早闭一般开始于出生后第一年,有些病例颅缝早闭可发生于出生前或出生时,但亦偶尔有颅缝未受累者。眼眶浅而眼球突出是Crouzon综合征诊断的最基本特征,在出生时至1岁时出现可以确诊,而在出生后头几年内逐步出现者要注意与其他疾病鉴别。在多数病例中,各骨缝可过早融合。在颅骨X线片上常见指状标记增多。
眼球突出为所有病人的特征。眼球突出是由于眶窝浅所致,继而使暴露性结膜炎或角膜炎的发生率高。在一些病例中可见眼袋松弛。外斜视是最常见的表现(77%)。近46%有视力低下,22%眼萎缩,7%失明。少见的症状还有眼球震颤、虹膜缺损、无虹膜、瞳孔不相等、瞳孔异位、小角膜、巨角膜、圆锥型角膜、白内障、晶状体异位、蓝色巩膜和青光眼。约50%的患者有腭外侧肿胀,有部分病例肿胀明显产生腭正中“伪裂”。由于Crouzon综合征患者的上颌骨发育不良,上颌牙弓前后距离变短、牙弓宽度变窄,狭小的牙弓使腭盖高拱(腭高度正常)。多数患者存在单侧或双侧后牙反合、上颌牙齿拥挤。上颌第一磨牙异位萌出,前牙开合。表现为上颌前突、下前牙拥挤、单根牙发育不良、上切牙牙冠短小及前磨牙形态异常畸形等。部分病人存在频发头痛,或癫痫及智力缺陷。
传导性听力缺陷和外耳道闭锁也较常见,伴鼻中隔偏曲、茎突韧带钙化等。颈椎异常在某些病例也可见(C5或C6融合及C2~C3~C5~C6融合)。桡骨头过度松弛,肘外翻,有些患者存在缺指(趾)畸形。严重病例因气管支气管软化症可发生气管阻塞和窒息。
Crouzon综合征应与单纯性颅缝早闭、Apert综合征、Pfeiffer综合征、Saethre-Chotzen综合征和Jackson-Weiss综合征鉴别。
Saethre-Chotzen综合征
本综合征呈常染色体显性遗传,主要表现为颅缝早闭。现已查明,本征主要与碱性螺旋-环袢-螺旋(basic Helix-Loop-Helix)转录因子基因(TWIST基因,定位于7p21)突变相关。TWIST基因突变可影响蛋白质的结构稳定、二聚化和DNA结合三个方面的功能而引起颅缝早闭。Saethre-Chotzen综合征的分子病因可分为下列四种情况:①TWIST基因部分或完全缺失;②TWIST基因的失活性突变;③FGFR2或FGFR3基因突变;④其他遗传性变异。
TWIST是成骨细胞分化的重要调节因子。TWIST基因的单倍体功能不全(Y103突变)可使成骨细胞的增殖和分化功能发生改变,同时也使成骨细胞易于凋亡,提示TWIST具有抗成骨细胞凋亡作用,但这些作用与Saethre-Chotzen综合征的表型特征有何联系不明。
从病人头颅骨分离的成骨细胞,突变基因表达的TWIST mRNA和蛋白减少,而细胞生长加速,ALP和1型胶原表达明显增加。在体外实验中,TWIST基因突变成骨细胞形成矿化结节加速,其合成胶原的能力显著增强,但骨钙素表达下降。多数病例有尖颅、面部畸形、发际下移、突眼、鼻中隔歪曲、三角耳及指(趾)畸形等,一些病例伴耳聋(传导性和感觉神经性)。TWIST基因突变所致的表型变异极大,在Dollfus报道的家系(Q28X)中,仅25%的病人有颅缝早闭,而上睑下垂者多达93%,其他表现也差异很大。造成表型差异的原因可能很多,如突变类型,优势负性作用及相关因子的作用等。此外,本综合征还可与其他颅缝早闭类型重叠发生。
临床诊断较容易,但应与Apert综合征的变异型及其他引起颅缝早闭的综合征鉴别(TWIST基因突变也可导致另一些颅缝早闭的表型综合征,如Baller-Gerold综合征)。TWIST基因诊断方法很多,如FISH和Southern杂交等。除TWIST突变外,FGFR3突变也可引起Saethre-Chotzen综合征(如FGFR3的P250R突变)。因此,在进行基因诊断时,可先进行FGFR3和TWIST筛选,如为阴性,可用FISH寻找缺失位点。
