
骨质硬化症分的几个类型
在第一届国际骨质硬化症研讨会议上,根据临床表现和基因分型,将骨质硬化症分为4型。
婴幼儿恶性型
婴幼儿恶性型骨质硬化症呈散发性发病,为常染色体隐性遗传。其病情严重,但许多病例的表现不典型,骨质硬化开始出现于新生儿期,有的患者可伴有Dandy-Walker综合征(先天性第四脑室中、侧孔闭塞等,是以第四脑室和小脑发育障碍为主的先天畸形)。患者为婴幼儿,一般在10岁以前死亡。约30%可存活到5~6岁,极少数可存活到20~30岁,患者的智力正常,骨骼硬化表现呈弥漫性分布。干骺端软骨钙化导致骨髓腔几近封闭,诸骨密度普遍增高,骨皮质与骨松质及骨髓腔隐约可辨,尺骨短小致前臂发育畸形,出现贫血,肝、脾和淋巴结的髓外造血和这些器官的肿大。而脑神经孔的形成亦受到影响,可出现视神经萎缩、眼球震颤、视乳头水肿、突眼和眼球运动障碍,常有颜面麻痹和耳聋,而三叉神经受损和嗅觉丧失则少见。患儿可有巨头、脑水肿和惊厥。骨骼密度增高,但脆性增加,易出现病理性骨折。由于造血系统的破坏,患者可因出血或感染而死亡。
基因分析表明,该类型患者有TCIRG1、CLCN7和GL基因突变。TCIRG1和CLCN7基因与氢离子泵及氯离子通道功能有关,而GL基因的功能则暂不明确。(补充:TCIRG1基因突变婴儿也可见脊椎椎体上、下部的硬化,呈“三明治”样外观。后面我附了一张我们发现该突变婴儿脊柱片,请您决定是否要用。)
成年良性型
Ⅰ型患者具有高骨量,与骨形成增加有关,并非为骨吸收下降所致。主要表现为颅骨、脊椎和骨盆硬化,但骨小梁的重建基本正常,骨的强度正常或增加,骨折少见。脑神经受损多见。该类型的基因分析表明为LDL受体相关蛋白5(LRP5)基因突变,该突变导致Wnt信号功能显著增强。因为Ⅰ型并非由破骨细胞的缺乏引起,因此有人建议将该型从骨质硬化症中分离出去。
Ⅱ型的临床表现较轻。贫血不严重,神经系统的异常也不常见。主要表现为颅骨和广泛性脊椎终板硬化。由于脊椎椎体上、下部的硬化更为明显,故可形成“三明治”样外观,而髂骨又可形成“骨中骨”外观。特点为反复发生的病理性骨折。部分患者因发生骨折才意外发现该病。此外还有骨骼疼痛、神经瘫痪和骨髓炎(常发生于拔牙后)等临床症状。该型的基因分析则为CLCN7基因单个等位点的错义或者移码突变等,但存在显著的外显不全。
中间型和伴肾小管性酸中毒的骨质硬化症
为常染色体隐性遗传。临床表现介于婴幼儿恶性型和成人良性型之间。是由CLCN7基因两个等位点突变引起。
伴肾小管性酸中毒的骨质硬化症
是常染色体隐性遗传。生存期较长,本型有肾小管酸中毒、脑钙化、智力下降、小脑畸形和牙咬合不全等特殊临床表现。骨中包埋有未吸收的钙化软骨岛。骨重建缺乏,骨结构紊乱,表现为骨皮质增厚和干骺端漏斗结构缺如。尽管骨密度升高,但骨的机械性能异常,易发生骨折。此型主要见于中东和地中海地区。是由碳酸酐酶Ⅱ基因突变引起碳酸酐酶Ⅱ缺陷症,进而导致破骨细胞吸收抑制。
常染色体隐性致命型的特征
常染色体隐性致命型以发育不良/身材矮小/肝脾大/脑积水/脑神经功能障碍/软骨内化骨为特征。本型遗传方式为常染色体隐性遗传,部分患者于出生后不久死亡,但不少患者可存活多年。主要临床表现为发育不良、身材矮小和肝脾大。因脑积水而头大,脑神经功能障碍,常伴耳聋和失明;部分有贫血和血小板减少以及反复感染等,但无智力障碍。
头颅穹隆和颅底均受累,特别是软骨内化骨的颅底更为严重。由于骨增生硬化而致神经孔(包括枕骨大孔)狭窄、颅板增厚、板障狭窄、蝶窦和鼻窦气化不良、上下颌骨还可有骨髓炎表现。脊柱椎体不均匀致密硬化,新生儿或婴儿早期,椎体前缘的血管沟或切迹明显;青春期由于椎体终极的增生硬化,椎体可呈“夹心面包”样改变。有时胸至腰段椎体内可见骨中骨。肋骨和锁骨增宽变厚,致密硬化,锁骨可有塑形障碍,肩胛骨亦明显硬化。管状骨普遍性骨硬化,骨松质、皮质和髓腔界限消失,骨构塑障碍使管状骨呈垒球棒样改变,骨端还可出现佝偻病样表现,如骨端膨大,有时尚可见骨膜增生以及病理骨折。骨盆可见致密硬化与稀疏透明相间的弧形带影,与髂翼外形一致,呈树木的年轮样改变。
中间隐性型病情隐缓伴矮身材/乳牙脱落延迟/骨折/脑神经受累
头颅可因脑积水而增大,颅盖骨直接受累较轻,颅底骨增厚、硬化,还可有鼻窦发育不良。脊柱椎体终板硬化,呈“夹心面包”样改变,还可见“骨中骨”表现。管状骨弥漫性骨硬化,正常骨结构基本消失,常易骨折并伴骨构塑障碍。髂骨可见年轮样改变。
常染色体显性遗传型的特征
常染色体显性遗传型以管状骨弥漫性硬化与皮质增厚和髓腔变窄为特征。81%的患者有明显症状,或因病理性骨折(78%)而被发现;可有轻度贫血,视、听力异常和牙齿不整等。病理性骨折后可出现局部骨畸形,愈合慢,少数并发骨髓炎(11%),2/3的患者伴脑神经受损,可引起视神经萎缩和失听等。患者智力正常。颅底骨,特别是前、中颅凹有明显的骨质硬化。椎体中部,即终板之间的骨松质BMD较低,整个椎体呈“夹心面包”样改变,还可见“骨中骨”。畸形严重者需要手术矫正。管状骨弥漫性骨硬化,骨骼、干骺区尤为显著,皮质增厚,髓腔变窄。一般无缺陷,可有骨折表现,有时可见“骨中骨”,或于干骺端出现“透明带”改变。骨盆可有不规则硬化区,常见于盆腔缘,髂骨翼区还可见致密与疏松相间的弧形带样改变。
隐性型合并肾小管酸中毒与器官损害
此型为常染色体隐性遗传,包括骨质硬化症、肾小管酸中毒和大脑钙化,患者可长期存活,部分患者有智力迟钝,某些患者缺乏碳酸酐酶(carbonic anhydrase)。主要临床表现与肾小管性酸中毒有关,如消瘦、肌张力低下、肌无力等,贫血较轻,可因病理性骨折引起骨畸形。Ⅱ型碳酸酐酶缺陷症主要见于地中海地区,偶见于土耳其及其他地区。Ⅱ型碳酸酐酶缺陷导致骨、肾和脑的损害,智力下降,生长发育延迟,本病呈常染色体隐性遗传,表现为骨质硬化、肾小管性酸中毒和大脑钙化,与其他原因所致的肾小管性酸中毒不同,本病患者无尿的浓缩功能障碍。
头颅普通X线可发现颅内钙化,CT或MRI扫描更可清楚显示出钙化的范围和部位。钙化可见于颅内任何部位,通常以基底核和脑室周围多发,病灶数目多。椎体呈弥漫性骨硬化,终板较椎体中部密度稍高,亦可有明显硬化。管状骨呈普遍性致密性硬化,尤其是骨骺和干骺区的密度更高,同时伴骨构塑(bone modeling)障碍,髓腔消失。骨的病变可逐渐改善,但易发生骨折。骨盆及其他骨均有弥漫性骨硬化表现。
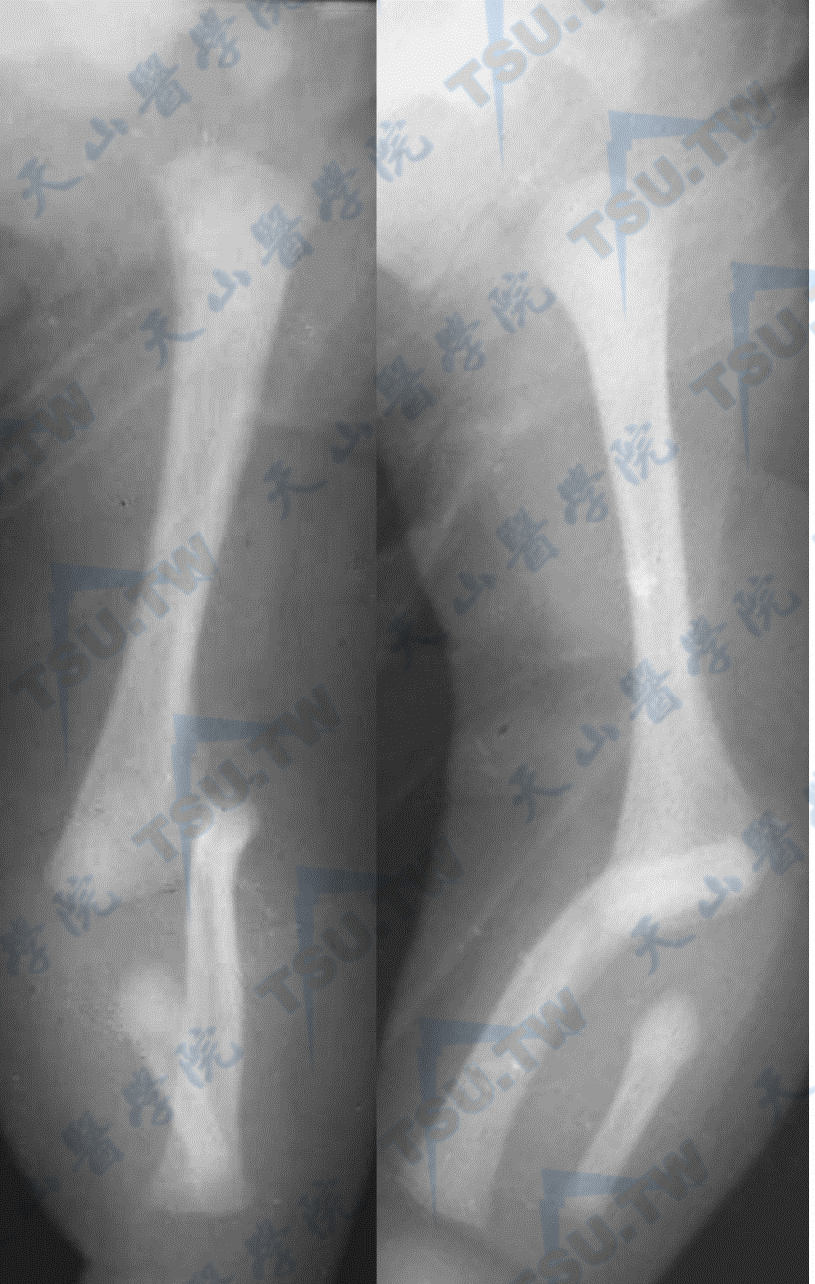

骨质硬化症
注:男性,3个月,骨质硬化症上肢及下肢平片。诸骨密度普遍增高,骨皮质与骨松质及骨髓腔隐约可辨,尺骨短小致前臂发育畸形
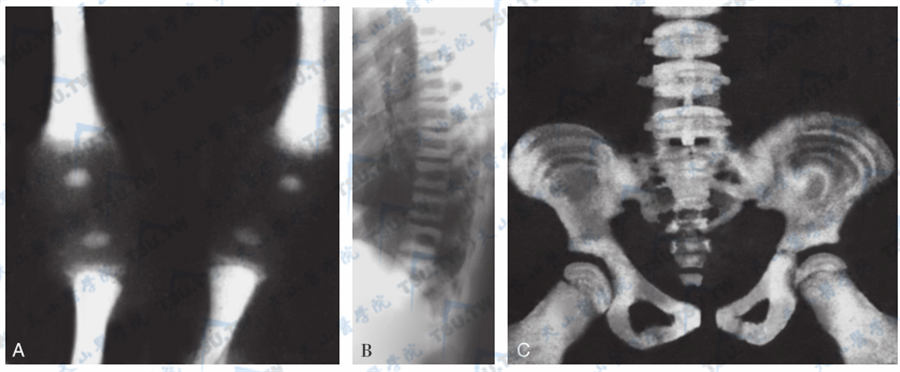
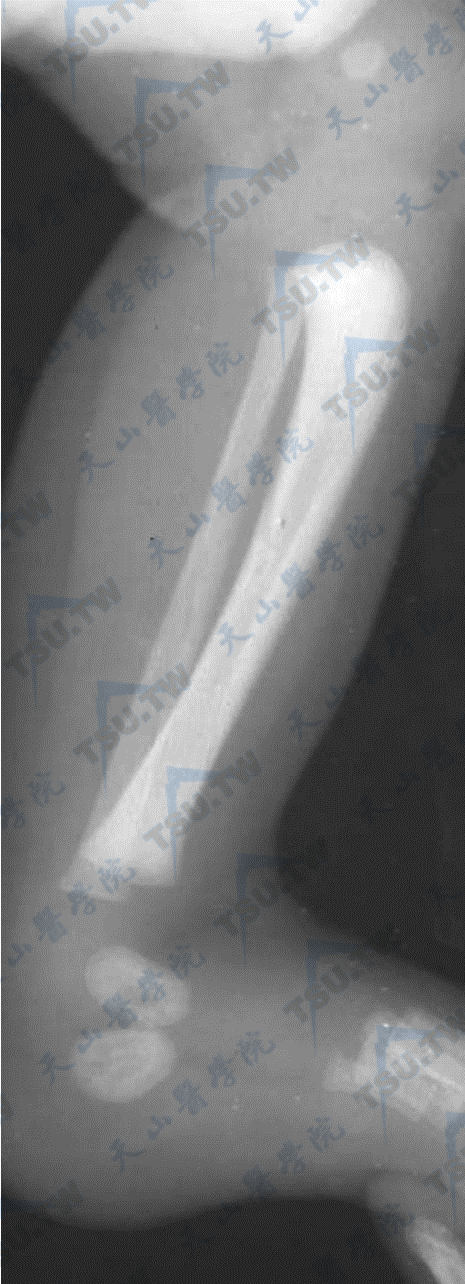
骨质硬化症
注:女性,新生儿。A:骨质硬化症下肢平片。股骨及胫骨密度甚高,呈粉笔样,皮质骨与松质骨及骨髓腔不能分辨。B:胸椎侧位。C:女性,9岁,胸椎及骨盆平片。诸骨密度增高,椎体以上下缘密度增高为甚,呈夹心饼样;骨盆诸骨较高密度带与相对稍低密度带交互排列,呈树轮状。
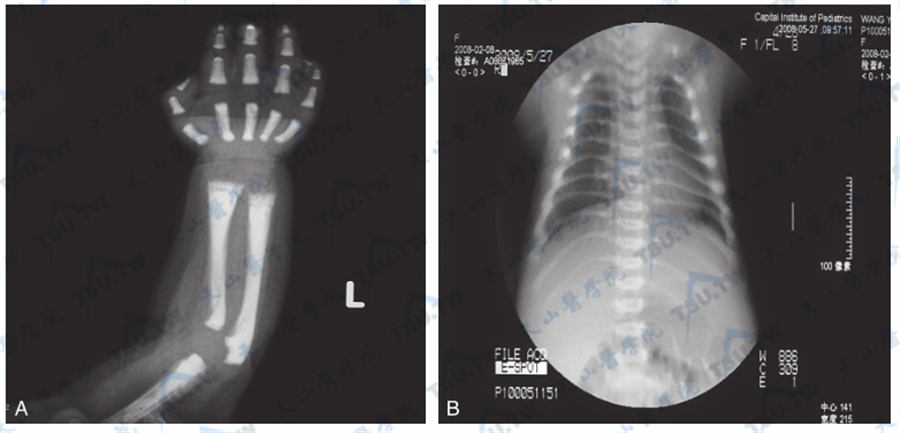
骨质硬化症的胸部与上肢X线照片
注:患儿,6个月龄,常染色体隐性遗传性骨硬化症(TCIRG1突变)
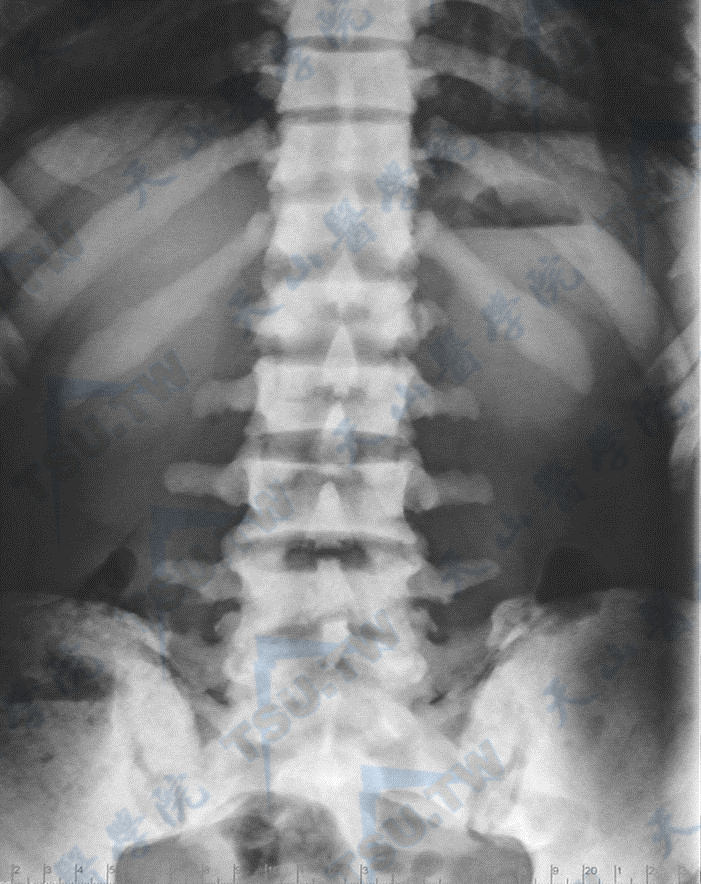
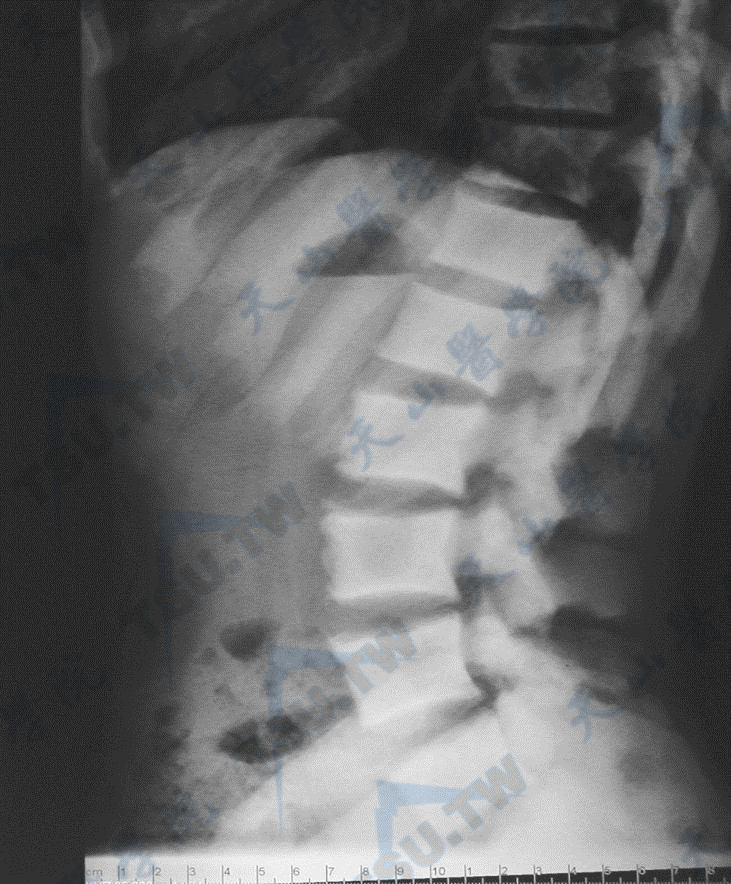
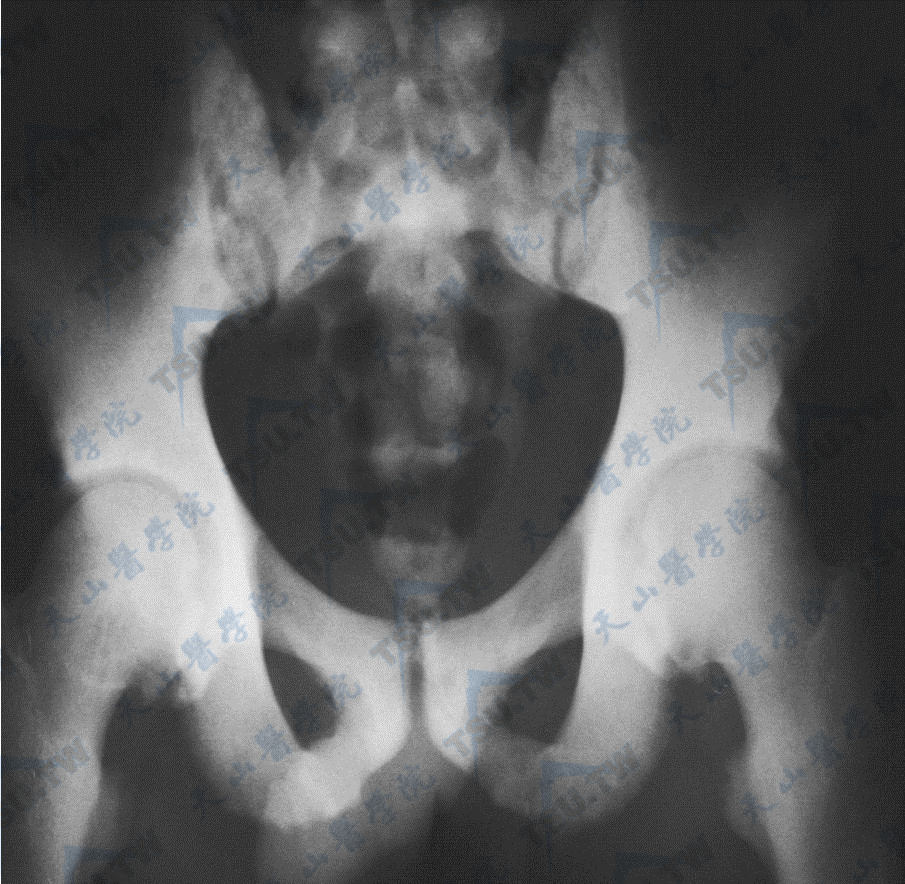
骨质硬化症的腰椎与髋部X线照片(1)
注:男性,37岁,全身骨骼硬化,散发,无家族史,没有筛查到已知基因突变
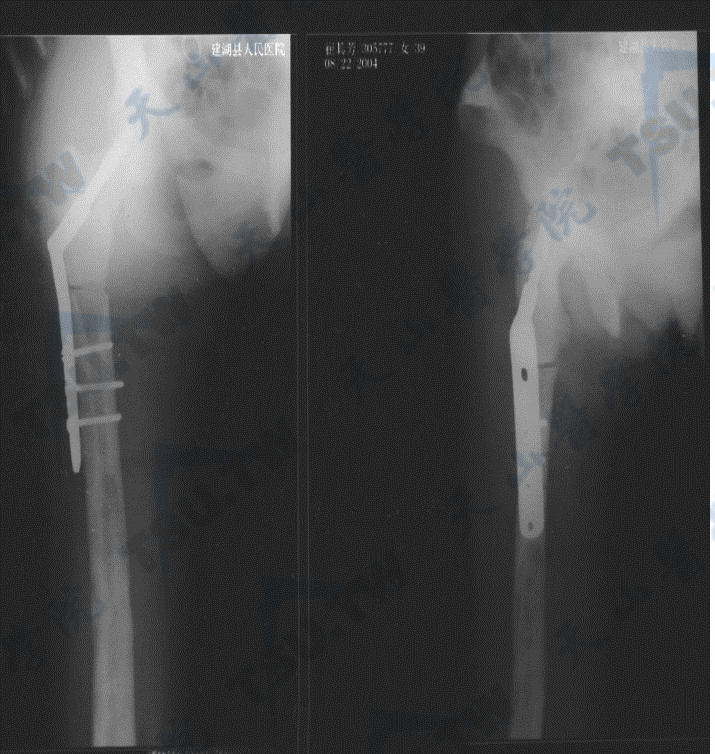
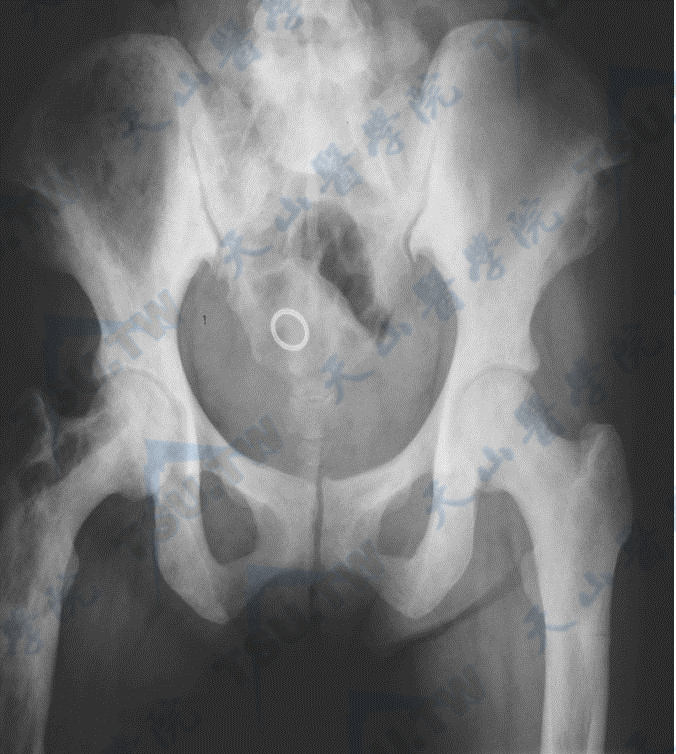
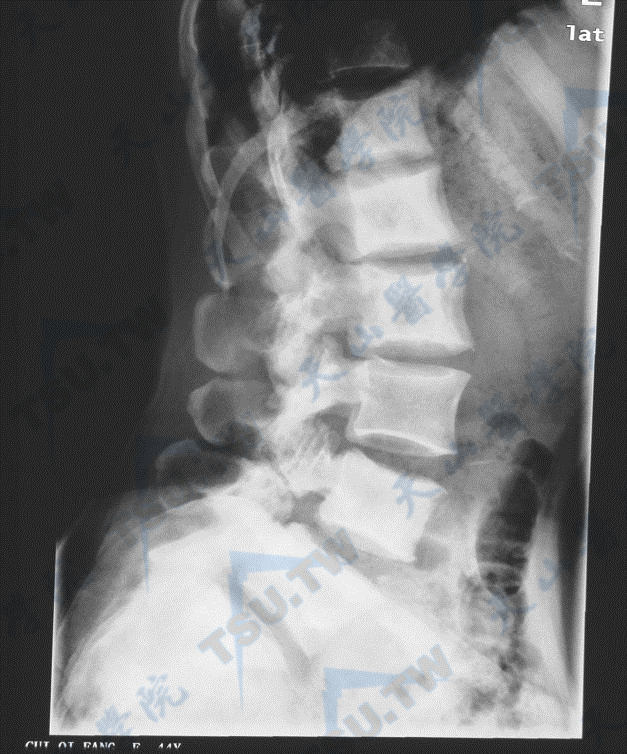
骨质硬化症的腰椎与髋部X线照片(2)
注:女性,51岁,多次骨折,全身骨密度高,血ALP 819U/L,未检出已知基因突变,可能为ADO-1或中间型骨质硬化症
