
硬化性骨病的病因复杂,分类方法不一。一般可根据临床表现、致病细胞(成骨细胞和破骨细胞)类型或细胞的信号途径进行分类。根据临床-病理类型,一般将骨质硬化症分为3种:①富含破骨细胞的骨质硬化症(osteoclast-rich osteopetrosis);②破骨细胞缺乏的骨质硬化症(osteoclast-poor osteopetrosis);③骨形成增强所致的骨质硬化症。Camurati-Engelman病和多发性干骺-骨骺点状硬化症(osteopoikilosis)是一种常染色体显性遗传的良性的干骺-骨骺硬化症,病因与转型生长因子β(transforming growth factor-β,TGF-β)信号异常有关;骨内膜骨质硬化(endosteal hyperostosis)、硬化性狭窄(sclerosteosis)、van Buchem病、高骨量综合征(high bonemass syndrome)和肢骨纹状肥大症(osteopathia striata)的发病均与调节破骨细胞分化的Wnt信号途径紊乱相关,而类花生酸信号因子突变引起Ghosal综合征(Ghosal syndrome)或厚皮性骨膜病(pachydermoperiostosis)。引起骨质硬化的其他突变因子有PLEKHM1、IKBKG(NEMO)、Kindlin-3、CalDAGGEF1、LEMD3和WTX等。
骨质硬化症分为三种临床-病理类型
按临床表现、累及的细胞类型或细胞信号途径可将骨质硬化症分为3种类型:①富含破骨细胞的骨质硬化症(osteoclast-rich osteopetrosis):主要与破骨细胞功能的相关基因突变有关;②破骨细胞缺乏的骨质硬化症(osteoclastpoor osteopetrosis):主要与破骨细胞分化相关的信号因子有关。③骨形成增强所致的骨质硬化症:有些骨质硬化症是由于骨形成增强所致,Camurati-Engelman病和脆性骨硬化(osteopoikilosis)与转型生长因子β(transforming growth factor-β)信号障碍有关;少见的隐性遗传性及显性遗传性骨质硬化症主要包括骨内膜骨质增生症(endosteal hyperostosis)、狭窄性骨硬化(sclerosteosis)van Buchem病、高骨量综合征和条纹状骨病(osteopathia striata)是调节骨形成的Wnt途径因子突变所致。Ghosal综合征与厚皮性骨膜病(pachydermoperiostosis)是由于类二十烷酸途径(eicosanoid pathway)功能障碍引起的。
大部分骨质硬化症具有遗传性,分为常染色体隐性和显性遗传性骨质硬化症两类,主要有4种类型(下表):
- 恶性骨质硬化(malignant osteopetrosis);
- Ⅱ型碳酸酐酶缺陷症(carbonic anhydrase deficiency type Ⅱ,CAⅡ);
- Ⅰ型常染色体显性遗传性骨质硬化症(autosomal dominant osteopetrosis typeⅠ,ADOⅠ);
- Ⅱ型常染色体显性遗传性骨质硬化症(autosomal dominant osteopetrosis type Ⅱ,ADOⅡ)。
从分子病因方面看,可能是由于破骨细胞刷状膜囊泡质子泵通道亚基基因(ATP6i基因或TCIRG1)突变所致,其动物模型为oc/ oc小鼠;致密骨发育不全症(pyknodysostosis)则是编码组织蛋白酶K(cathepsin K)的基因失活性突变所致,而Camurati-Engelmann病是因为骨形成增强引起的骨质硬化症,可能与TGF-β1基因突变有关。
人类骨质硬化症的分类(分型)
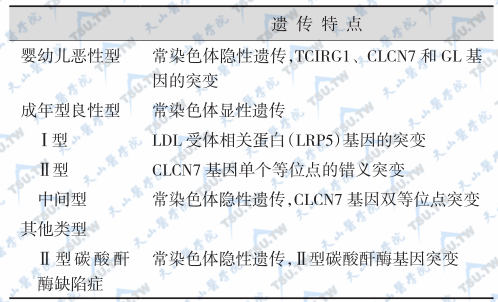
注:TCIRG1:氢离子泵H+-ATP酶;CLCN7:氯通道7;GL:灰色致死
凡引起破骨细胞功能障碍的因素都可能导致骨质硬化。OSTM1基因突变伴有中枢神经病变、视神经损害和生长发育障碍。临床表现为骨质硬化伴中枢神经病变、生长发育障碍、贫血、血小板减少和视神经损害。这些病例与经典的临床类型有别,可能属于一种新的骨质硬化症。另外,发现骨质硬化症患者的破骨细胞内过氧化物酶的分泌下降,仅为正常的30%,亦导致骨吸收减少。在动物试验研究中,导致破骨细胞吸收功能下降的因素很多。op/op鼠不能表达M-CSF,导致破骨细胞的早期分化缺陷;c-Src-/-小鼠因为M-CSF受体缺乏导致破骨细胞早期分化缺陷;PU.1敲除小鼠因PU.1(M-CSF受体基因转录子)缺乏亦导致破骨细胞早期分化缺陷。在RANKL基因敲除小鼠中,因为破骨细胞RANKL缺乏而导致骨质硬化,相反,护骨素转基因小鼠因护骨素过多而导致破骨细胞数量减少,产生骨质硬化;此外,在小鼠模型中,Mitf、c-fos、TRAF-6、c-src、gl/gl基因缺乏都可以导致骨质硬化。
遗传性骨质硬化症的病因分类
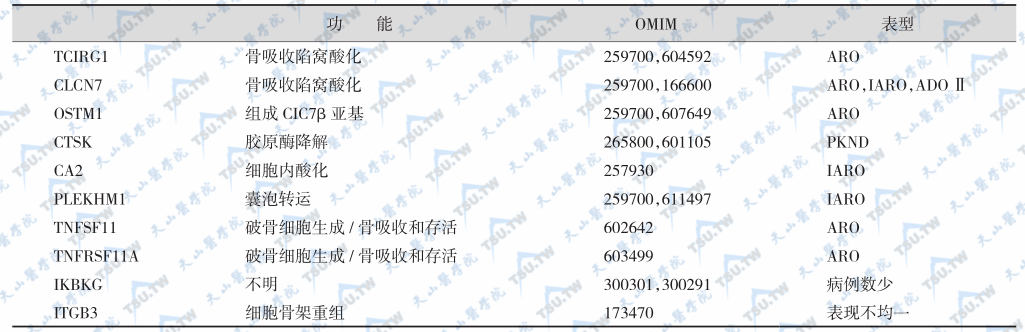
TCIRG1:T-cell,immune regulator 1,ATP,H+transporting lysosomal VO subunit A3,T细胞免疫调节子1-ATP酶-H+转运溶酶体VO亚基A3;CLCN7:chloride channel 7,氯通道7;OSTM1:osteopetrosis associated transmembrane protein 1,骨质硬化相关性跨膜蛋白1;CTSK:cathepsin K,组织蛋白酶K;CA2:carbonic anhydrase Ⅱ,碳酸酐酶Ⅱ;PLEKHM1:pleckstrin homology doncain containing,family M(with RUN domain)member 1,血小板-白细胞C激酶底物同源序列M家族成员1;TNFRSF11A:tumor necrosis factor(ligand)superfamily mencher11A(RANK),TNF超家族成员11A(RANK);IKBKG:inhibitor of kappa light polypeptide gene enhancer in B-cells,kinase gamma(NEMO),β细胞多肽基因增强子轻链κ抑制物,激酶γ(NEMO);ITGB3:β3 integrin,β3整合素。ARO:autosomal recessive osteopetrosis,常染色体隐性遗传性骨质硬化症;ADO:antosonal dominant osteoetrosis,常染色体显性遗传性骨质硬化症;IARO:intermediate autosomal recessive osteopetrosis,中间型常染色体隐性遗传性骨质硬化症;PKND:pycnodysostosis,致密性成骨不全症。
骨质硬化鼠模型基因分析
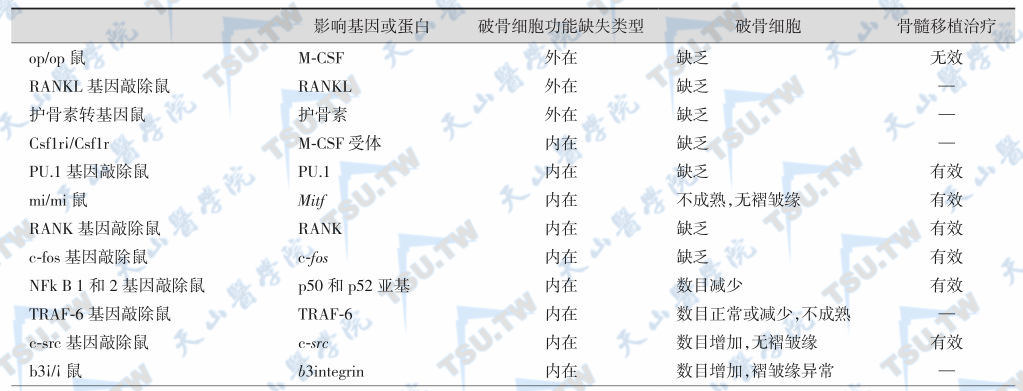

注:M-CSF:巨细胞集落刺激因子;CLCN7:氯通道7;ATP6i:氢离子泵H+-ATP酶;integrin:整合素;RANKL:核因子κB受体活化因子配基;Grey-lethal:灰色致死,GL。
(二)碳酸酐酶Ⅱ突变引起骨质增多伴肾小管酸中毒及脑钙化
破骨细胞碳酸酐酶Ⅱ缺陷症呈常染色体隐性遗传。破骨细胞胞质内的碳酸酐酶Ⅱ将二氧化碳和水合成为碳酸,碳酸可以分解为氢离子和碳酸氢离子,前者通过氢离子通道转运到破骨细胞膜褶皱区的吸收腔隙,完成破骨细胞的骨吸收过程。碳酸酐酶Ⅱ基因的缺乏导致氢离子的生成减少,进而影响破骨细胞的吸收功能,引起骨质增多。碳酸酐酶Ⅱ基因缺乏有特殊的地域相关性。该类型的患者较少见,在骨质硬化症患者中的比例少于5%。
破骨细胞碳酸酐酶Ⅱ基因突变伴有肾小管酸中毒及大脑钙化的骨质硬化症患者存在碳酸酐酶Ⅱ(carbonic anhydrase Ⅱ)的缺陷,其特点是骨硬化、肾小管性酸中毒、脑组织钙化和智力缺陷。此酶基因缺陷的类型较多,一般为点突变所致(如第6号内含子5′端点突变或外显子7的单碱基缺乏)。石骨症伴颅骨干骺端发育不良者,破骨细胞胞质内的囊泡存在着囊泡质子泵表达缺陷。破骨细胞通过酸化破骨表面而溶解基质中的无机盐,分泌溶酶体酶和非溶酶体酶而降解有机质。破骨细胞以胞吐方式使浆膜突出,形成刷状膜,后者使破骨细胞膜与骨表面的接触面扩大,接触更为紧密。如果破骨细胞的这些功能异常,均可导致骨吸收功能下降,引起骨质硬化症。婴幼儿型骨质硬化症患者的破骨细胞并不缺乏,但破骨细胞缺少刷状膜,此与ATP6i(TCIRG1)基因敲除鼠的表型类似。ATP6i基因编码ATP酶的a3亚基突变(如缺失/插入突变)导致ATP酶蛋白被截短,功能丧失,引起婴幼儿型骨质硬化症,但其发生机制未明。
以上研究表明,有多种基因突变可引起骨质硬化症。此外,有些骨质硬化症患者的破骨细胞缺陷表现为无骨吸收功能,虽然细胞膜含有降钙素受体和玻连蛋白(vitronectin)受体,但细胞不能表达CD44或表达量很低。
(三)氢离子泵或氯离子通道缺陷导致骨吸收障碍
骨细胞氢离子泵功能缺损综合征亦呈常染色体隐性遗传。氢离子泵H+-ATP酶(TCIRG1,又称为ATP6i或OC116)基因突变导致破骨细胞细胞膜上的氢离子泵A3亚单位结构异常,因此,虽然氢离子泵表型正常,但是无正常的转运功能,从而氢离子的转运受制,导致破骨细胞的吸收功能下降。
通过常染色体隐性和显性两种方式遗传。破骨细胞细胞膜上有很多氯离子通道(CLCN7),具有转运蛋白、参与膜内陷和跨细胞转运的作用。其中有一部分特殊的氯离子通道,称为CLIC5,保持氢离子泵转运过程中细胞内的电平衡,CLIC5和H+-ATP酶比例为1∶2000。氯离子通道基因的突变导致继发性的氢离子转运异常,从而影响破骨细胞的吸收功能。在常染色体隐性遗传中,CLCN7基因的突变发生于两个等位基因,而在显性遗传中,其突变只表现为单个等位基因的错义突变。临床常见的是CLCN7基因突变导致的常染色体显性遗传的2型(ADO-Ⅱ)]。但是,通过对CLCN7突变检测提示,ADO-Ⅱ病例中,CLCN7基因突变存在明显的外显不全,即同一家族中相同杂合突变的个体,有的不发病,其中机制不明确,是否存在修饰基因或者CLCN7突变个体DNA甲基化等尚待阐明。
(四)TCIRG1突变引起婴幼儿型骨质硬化症
ADOⅡ是骨质硬化症中的最常见类型,致病基因位于16p13.3。Sobacchi等发现,约50%的常染色体隐性遗传石骨症(autosomal recessive osteopetrosis)患者存在TCIRG1基因突变,突变类型包括基因缺失、插入、无义突变、错义突变和剪接变异等。Cleiren等发现,氯通道基因CICN7的纯合子突变可引起婴幼儿型骨质硬化症(infantile osteopetrosis)。ADOⅡ似乎是由于杂合子突变基因的优势负性作用(dominant negative effect)所致,而CICN7的失活性杂合子突变因无优势负性作用特点,故只有纯合子才发病,此时只有通过基因突变分析才能将两者鉴别开来。有人认为,CICN7氯通道病就是ADOⅡ的一种亚型。
(五)CSF1与OPGL突变引起骨质硬化症
无牙(toothless,t1)鼠的CSF1基因突变引起骨质硬化症。无牙鼠是由于自发性常染色体隐性遗传性CSF1基因突变所致,是研究CSF-1功能的良好动物模型,联合OPG基因缺失(op/op)鼠动物模型,可深入阐明骨质硬化和骨质疏松的发病机制。近年对骨质硬化鼠动物模型的研究发现,缺乏粒细胞-巨噬细胞集落刺激因子(GM-CSF)的小鼠可出现自发性骨硬化。GM-CSF可刺激骨吸收,促进破骨细胞的前身细胞转化成为成熟的破骨细胞。破骨细胞能分解胶原蛋白,吸收骨基质游离骨盐。如果CSF的基因突变使CSF无生物活性,则使造血前体细胞发育缺陷,引起破骨细胞发育障碍、骨吸收损害及石骨症。M-CSF基因突变鼠的骨质硬化在用重组鼠GM-CSF (rmGM-CSF)和IL-3治疗后,骨质硬化病变得到逆转,也反证了骨质硬化的发生与GM-CSF和IL-3缺乏或活性下降有关。
另一方面,近年来相继克隆出肿瘤坏死因子(TNF)家族中的护骨素(osteoprotegerin,OPG)及其配体(RANKL)。这些细胞因子、受体及其配体组成骨代谢的局部调节系统,主要通过调节破骨细胞的形成、活性与凋亡而调节破骨细胞的功能及成骨-破骨耦联过程。一些研究表明,骨质硬化可能是RANKL和M-CSF的产生、受体活化、细胞内传导途径和核受体活化障碍所致,RANKL基因突变亦发生严重的骨质硬化,破骨细胞缺乏。Klotho基因突变鼠(k1/k1)有骨质硬化表现,其病因与骨组织表达的OPG(为野生鼠的2倍)升高有关。另一方面,OPG基因被破坏后,小鼠有骨质疏松表现。
