确立McCune-Albright综合征
根据骨纤维结构不良/皮肤café-au-lait斑/性早熟确立McCune-Albright综合征(MAS)诊断。临床可根据下列4条确定MAS的诊断:
- 有骨损害、皮肤色素沉着和性早熟3大主征(MAS三联征);
- 有骨纤维结构不良的X线表现和皮肤café-au-lait斑;
- 伴有内分泌(主要是青春期发育提前)或非内分泌异常的年轻(30岁以下)患者,如有TSH依赖性甲亢,ACTH依赖性Cushing综合征或非GnRH依赖性性早熟及相应的激素与生化代谢变化,则更支持本病的诊断;
- Gsα基因突变。
本病的骨骼呈纤维结构不良改变,纤维组织丰富,新生的骨小梁被挤压。纤维结构不良与GNAS突变直接相关,引起成骨细胞分化障碍和骨吸收增。用变性梯度凝胶电泳(denaturing gradient gel eletrophoresis)和特异性对耦联微量核苷酸杂交(allele-specific oligonucleotide hybridization)方法可分析Gsα基因的R201C 和R201H突变,从而为MAS提供分子病因诊断和治疗依据。因患者外周血液白细胞Gsα亚基均未见异常(体细胞突变,somatic mutation),所以应取病变(骨骼或皮肤)组织标本才能得到阳性结果。
但MAS的分子病因诊断有赖于Gsα基因的突变分析。
MAS骨纤维结构不良与其他骨病鉴别
一、Paget骨病
MAS的骨病不典型时,易与Paget骨病相混淆,但Paget骨病无性早熟,亦无皮肤café-au-lait斑,而血ALP明显升高。
二、神经纤维瘤病
累及骨骼,常并有皮肤咖啡斑,可与MAS类似。但神经纤维瘤病有皮下结节或软性包块改变及多发性神经纤维瘤,不合并内分泌异常,亦无性早熟。
三、肿瘤所致的低磷血症性骨质软化症
用MAS患者体内表达突变型Gsα亚基的细胞(MAS细胞)做实验,发现引起MAS患者低磷血症的体液因子不同于肿瘤相关性低磷血症性骨质软化症,这种因子的性质未明,其特点是抑制肠磷吸收而不抑制肾小管磷的重吸收。
四、骨纤维结构不良症
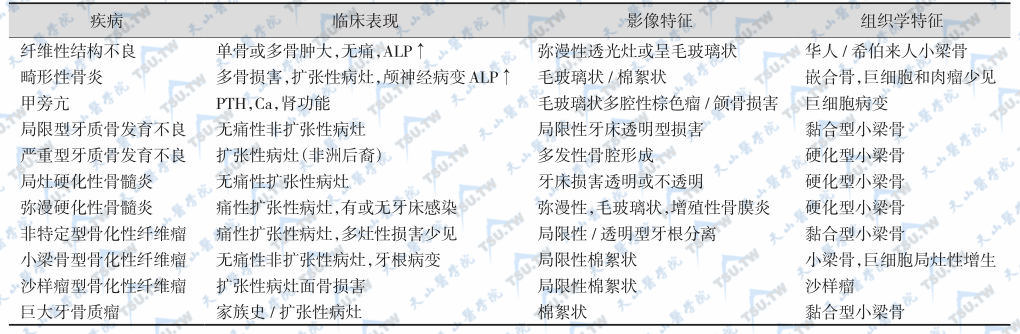
纤维发育不良症(fibrous dysplasia)和骨纤维发育不良症(osteofibrous dysplasia)为骨结构异常的良性疾病,在组织学上和临床表现方面,有时很难鉴别。据Sakamoto等报道,纤维发育不良症(7例)均存在Gsα亚基Arg201密码子突变,而骨纤维发育不良症(7例)均无突变。因此,Gsα亚基基因的突变分析有助于两种骨损害的鉴别。颅面纤维性骨病变(fibro-osseous lesions of the craniofacial complex)是临床鉴别诊断的重点,引起此类骨损害的原因很多(下表),当疾病处于在前阶段时,鉴别相当困难。
五、进行性骨化性纤维发育不良症
颅面纤维性骨病分类

注:NOS:Not otherwise specified,非特定型
各种纤维性骨病变的异同点比较

此病应与进行性骨化性纤维发育不良症(fibrodysplasia ossificans progressiva,FOP)鉴别,但后者无骨骼畸形表现,亦无炎性肿瘤样骨膨胀,骨化的方式为膜内成骨而非软骨内成骨。POH与MAS鉴别的要点是:MAS中的AHO的异位骨化呈进行性发展,由皮肤、皮下组织逐渐累及骨骼肌,甚至韧带组织。POH可合并MAS,一些POH患者的Gsα基因突变使Gsα亚基失活、Gsα亚基下降。看来,Gsα的失活性突变是引起骨骼肌和深部结缔组织骨化的分子病因(如Q12X突变)。可以认为,POH 和AHO分别为异位骨化临床表现谱的两种极端现象,典型POH无MAS的临床表现,非典型POH则伴有AHO和(或)短指(趾)畸形、肥胖及PTH抵抗综合征。因此,从分子病因上看,POH和AHO又是同一基因突变疾病的两种表现型,其中骨外骨化为Gsα亚基缺陷的表现型形式之一。
纤维性骨病变的临床表现、影像与组织学特征
六、多骨溶解-骨质增生综合征
Kantaputra等报道一例该病例,表现为多个长骨和骨盆有扩张性骨溶解、颅骨骨质增生、笼状胸、房间隔缺损、心脏扩大、隐睾和精神异常等,其临床表现与青少年型Paget骨病有许多相同之处,可能为常染色体隐性遗传。多骨溶解-骨质增生综合征(polyosteolysis/hyperostosis syndrome)分为4种病理类型:①扩张性骨溶解;②扩张性单纯性骨溶解而无骨扩张;③单纯性骨扩张骨而无骨溶解;④骨质增生。
七、异位骨化
异位骨化(heterotopic ossification)具有进行性骨纤维结构不良症、进行性异位骨化和AHO等的共同特点,均属于遗传性骨代谢疾病,但它们的骨化机制、组织病理、异位骨化部位和发展与预后各不相同,应注意鉴别。
八、盘状皮骨瘤
盘状皮骨瘤(platelike osteoma cutis,POC)为POH的一种亚型。如自幼出现皮肤和皮下脂肪组织骨化,并逐渐累及骨骼肌(面部、头部、眼眶周围等),提示为AHO或异位骨化伴有AHO。鉴别的唯一方法是进行Gsα基因的突变分析。Yeh等报道1例POC患者(女性)伴先天性皮肤和皮下组织骨化,患者Gsα基因的第7号外显子缺失4bp(杂合子,阅读框架移位,终止密码子提前出现,伴13个氨基酸残基被替代),这再次证明Gsα基因突变为POH的分子病因。
九、Mazabraud综合征
主要临床特点是骨的纤维性发育不良和骨骼肌黏液瘤病。Faivre等报道2例Mazabraud综合征患者,皮肤有Café-au-lait斑及多发性结节性甲状腺肿。到目前为止,已有37例Mazabraud综合征报道,其中6例伴有AHO,但患者无Gsα基因突变。
性发育提前与其他性早熟鉴别
女性真性性早熟
系完全性同性性早熟。由于性腺轴提前发育、青春期过早来临,性成熟过程按正常青春期顺序进行;下丘脑-垂体-性腺轴功能建立后,有排卵性月经周期和生育力。主要有3种:①特发性性早熟:是小儿真性性早熟的常见原因,有家族发病倾向,常染色体隐性遗传。多数在4~8岁间发病,阴毛随同外生殖器的发育而出现。月经周期由不规则逐渐变为规律并出现排卵。②中枢神经系统疾病所致的真性性早熟:常见的肿瘤有松果体瘤、视神经胶质瘤、下丘脑错构瘤、鞍上畸胎瘤、神经纤维瘤、星形细胞瘤、室管膜瘤或蛛网膜囊肿。除性征发育外,同时伴随颅内疾患的其他相应症状,如多饮、多尿、发热、肥胖或过度消瘦、精神异常、智力发育迟缓、头痛、呕吐、惊厥、肢体瘫痪及视力障碍等。③其他原因引起的真性性早熟:见于先天性肾上腺皮质增生症如11-羟化酶和21-羟化酶缺陷症患者经糖皮质激素或同时盐皮质激素治疗,血浆ACTH水平受抑制,肾上腺产生的性腺甾体类减少。但由于此前延误诊断和治疗,患者骨龄提前,如已达到青春期启动的骨龄界限值,患者可出现下丘脑-垂体-性腺轴功能的激活,引起性早熟。
女性假性性早熟
系不完全性同性性早熟,性腺或肾上腺来源的雌激素或外源性雌激素过多刺激靶器官,造成第二性征发育和月经来潮。因未建立正常下丘脑-垂体-性腺轴功能,故无生育力。女性假性性早熟可见于以下情况:
- 卵泡囊肿、卵巢肿瘤(颗粒细胞瘤、泡膜细胞瘤等)和肾上腺女性化肿瘤是女性假性性早熟的最常见原因,雌激素来源于肿瘤或分泌的雄烯二酮在腺外转化而来。B超检查有助于卵泡囊肿与实质性卵巢肿瘤鉴别。
- Peutz-Jeghers综合征的主要病变为黏膜皮肤色素沉着、消化道息肉瘤和性索瘤。因肿瘤分泌雌激素而出现不完全性性早熟,偶尔伴支持细胞-间质细胞瘤。
- 少数幼年和少年期甲状腺功能减退可出现性早熟,表现为乳腺发育、小阴唇增大、阴道黏液涂片可见雌激素影响的变化。一般无阴毛生长,部分患儿身材矮小,骨龄常落后于实际年龄。卵巢内可出现单个或多个小囊肿,可伴有阴道不规则流血。
- Russell-Silver综合征有身材矮小、骨龄延迟和头颅及面骨发育异常,表现为倒三角形脸,口角向下,身材明显不对称,指、趾骨并指(趾)或第5指(趾)内弯、短小畸形。34%的患儿有性早熟(智力大多正常)。
- 外源性雌激素包括含雌激素的药物(如口服避孕药及其他含雌激素的食物)等均可引起假性性早熟。
男性中枢性性早熟
特发性真性性早熟可开始于性发育前的任何年龄,性征发育的次序与正常儿童一样,男性先有睾丸和阴茎肥大,继之阴囊皮肤皱褶增加伴色素加深,甚至有精子生成。血清LH、FSH增高,伴性激素增高。如连续多次采血,可发现LH呈脉冲式分泌。
中枢神经系统疾病所致性早熟多由器质性脑部病变所致,包括下丘脑肿瘤、感染、囊肿、脑积水和脑外伤等,其性发育过程与特发性真性性早熟相似。两型的区别在于后者不能查出相应的器质性疾病。
男性外周性性早熟由于下丘脑GnRH和垂体促性腺激素以外的雄性激素刺激引起,包括促性腺激素(分泌LH或hCG的肿瘤)或性激素(先天性肾上腺皮质增生症、肾上腺或性腺肿瘤)的异常分泌,或影响性激素产生的基因突变所致。主要见于:①分泌促性腺激素的肿瘤。②雄激素产生过早过多。③睾酮中毒症(testotoxicosis)。④原发性甲状腺功能减退伴性早熟。⑤外源性性激素。
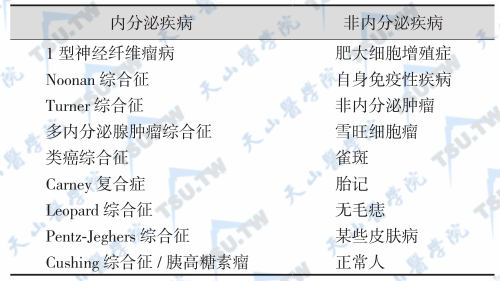
café-au-lait斑多种疾病鉴别
皮肤Café-au-lait斑并非MAS的特有表现,café-aulait斑亦见于MEN、类癌综合征、Cushing综合征、McCune-Albright综合征、Noonan综合征、Turner综合征、多发性神经纤维瘤病、Carney复合症、Leopard综合征、Pentz-Jeghers综合征、胰高糖素瘤、Pentz-Jeghers综合征和肥大细胞增殖症。许多非内分泌疾病也可伴有café-au-lait斑,如肥大细胞增殖症、自身免疫性疾病、雪旺细胞瘤(schwannoma)和正常人。此外,café-au-lait斑必须与雀斑、胎记(birthmarks)、无毛痣或一般性皮肤损害鉴别。Carney复合症患者有色素痣和斑点状色素沉着,类似于McCune-Albright综合征和von Recklinghausen病(多发性神经纤维瘤病)的café-au-lait斑,但Carney复合症的皮肤颜色较浅、面积小且多在面部中央密集,通常随增龄而消退。
NF-1的临床特征为牛奶咖啡斑、神经纤维瘤、腋窝和腹股沟雀斑、虹膜错构瘤、视神经胶质瘤及骨发育障碍,通常好发于躯干,随增龄而有增多、扩大的趋势;腋窝或腹股沟雀斑也是本病皮肤表现的特征之一。Pentz-Jeghers综合征多见于儿童和青少年,是一种常染色体显性遗传病,患者有胃肠道多发性错构瘤样息肉,手足皮肤及口腔黏膜的色素沉着,故又称为皮肤黏膜黑斑息肉病,息肉最常见于小肠,不具备Carney复合症的主要特点(黏液瘤和其他内分泌肿瘤)。此外,café-au-lait斑还需与多种系统性疾病、自身免疫性疾病、皮肤病、普通雀斑及色素痣鉴别。
伴有café-au-lait 斑的临床疾病