MAS为G蛋白病中的一种,其病变广泛,大多数情况下为一种良性骨病综合征,少数患者可出现肢端肥大症、Cushing综合征或高泌乳素血症。心肌病和由此引起的心律失常往往是猝死的重要诱因。灶性骨损害呈膨胀性扩大可导致病理性骨折,颅底骨增生硬化常导致视神经萎缩,面骨畸形引起五官粗陋。性早熟虽有自限性特点,但多数遗留身材矮小和肥胖等后遗症。
灶性骨病由结缔组织/交织骨/软骨结节组成
病变自骨髓腔向骨皮质膨胀性侵犯,导致骨皮质变薄,可有液化、囊变、出血和结节内骨化,形成局灶性畸形,累及骨承重部位可导致跛行和病理性骨折。
病变可累及全身骨骼,根据病变性质可分为单发型和多发型两种。单发者以股骨、胫骨和肋骨最常见,脊柱和骨盆少见。30%累及颅面骨,以上、下颌骨和颅骨顶部为主。颅底骨质增生硬化常压迫脑神经;波及视神经时,导致视神经萎缩。面骨过度增生,使面容不对称,鼻窦闭塞。脊柱、骨盆和四肢长骨的病变导致骨痛、骨畸形及骨折。多发性者累及身体双侧或以一侧为主,常见于下肢、股骨、胫骨和骨盆,较少累及的部位为肋骨、颅骨和颅底。
当早期发现疑似小病变,或因病变内交织骨含量少,平片表现无特异性时,CT可显示微量钙化小灶交织骨;临床怀疑为本病,平片未发现骨骼病灶,应选用CT/MRI进一步检查头颅、脊柱及骨盆,有利于早期诊断。
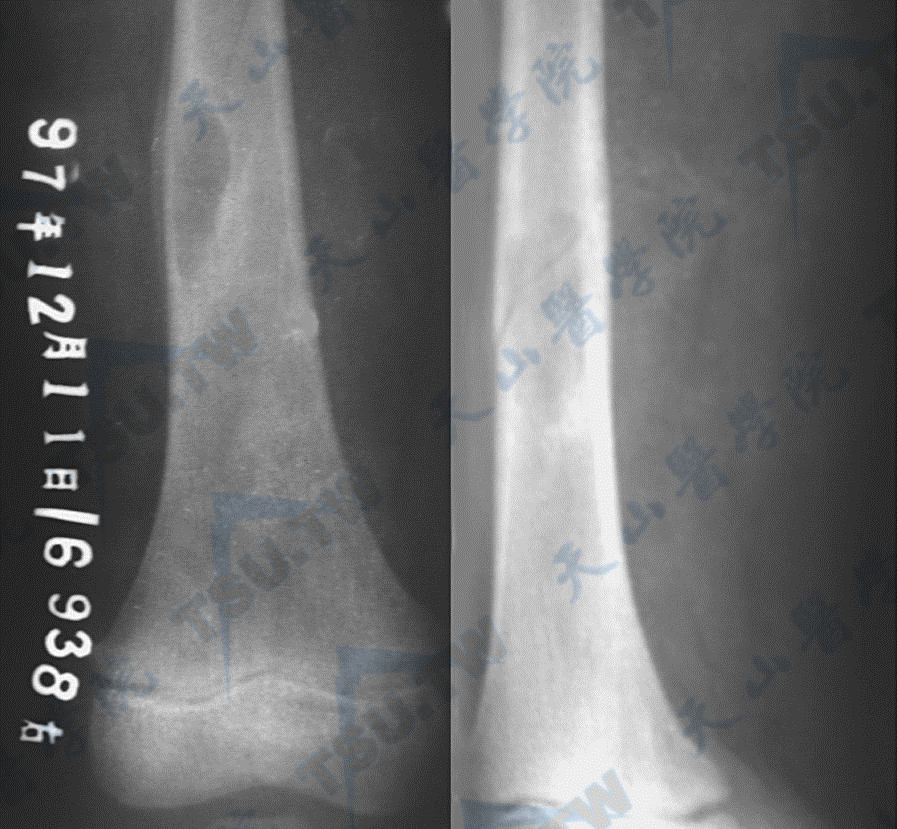
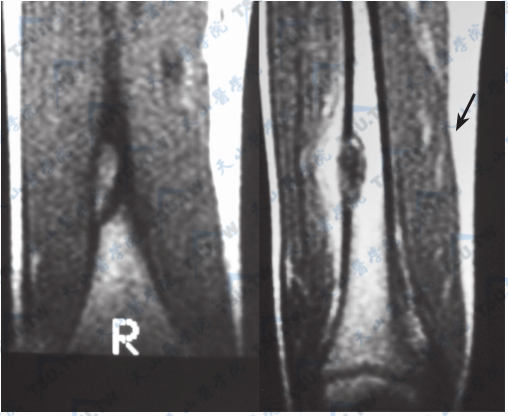
Albright综合征的骨纤维结构不良(MRI)
café-au-lait斑提示MAS
皮肤色素沉着的形状不规则,常呈小片状分布,多见于背部,亦可见于口唇、颈背、腰臀部和大腿等处。出生时,色素斑可不明显,随年龄的增长或阳光曝晒而加重、变深,受累面积扩大。有些患者的café-au-lait斑可能很浅,不容易被发现,或者因为café-au-lait斑为灰色甚至粉红色而漏诊。皮肤病变的外形与骨病变的多少有关。如色素沉着边缘清晰,一般仅单一骨受累;若边缘呈地图状,一般为多部位骨受累,但café-au-lait斑的病因可能是相同的。
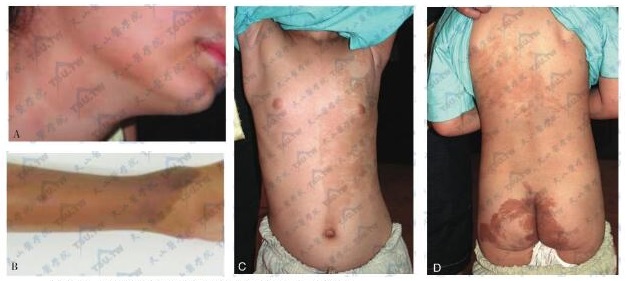
Albright综合征的皮肤café-au-lait斑
注:A:颈部和下颌皮肤café-au-lait斑;B:手臂皮肤café-au-lait斑;C和D:女,4岁,患者的正面及背面像。示乳房开始发育,胸、腹部、背部及臀部均有深浅不一的褐色斑,不高出皮肤,胸、腹部皮肤色素沉着呈偏侧性分布。
发生咖啡斑(café-au-lait macules)的原因仍不清楚,Gsα活化性突变导致皮肤色素细胞的MSH受体自动激活,皮肤色素浓集与沉着。但是,皮肤色素细胞还可表达许多其他G蛋白耦联受体和细胞因子受体,其中有些调节因子具有拮抗色素沉着作用,这些因素综合平衡的结果是咖啡斑的颜色深浅不等。此外,不表达Gsα活化性突变的皮肤细胞色素正常,这可能是边界清晰的重要原因。一旦出现,色素斑很少受到外界因素或药物的影响,故可经久不褪。如上所述,咖啡斑的主要特征是:①多为深棕色或浅咖啡色,但亦可为浅蓝、浅灰甚至粉红色,色素斑内的色泽基本一致,且边界清晰;②不突出表面,无自觉不适;③与生俱来或出生后发生,色素斑不消退但可加深。咖啡斑可见于许多临床情况,缺乏诊断绝对特异性。咖啡斑应首先与“胎记(birthmarks)”鉴别,后者亦可呈咖啡色,但较细小,且一般边界不清。遇有咖啡斑时,应首先想到McCune-Albright综合征、1型神经纤维瘤病(NF-1)、Noonan综合征、Carney复合症等可能。
将MAS患者皮肤café-au-lait斑处的皮肤细胞进行体外培养和实验,发现黑色素细胞中的Gsα亚基有突变,细胞内cAMP升高,酪氨酸激酶表达增加,说明café-au-lait斑的原因与黑色素细胞Gsα亚基活化性突变有关。
性发育提前属于特殊的假性性早熟
女性青春期发育启动后,性征和性功能发育的顺序是:体型改变→骨盆增宽→乳腺发育→阴毛和腋毛生长→月经来潮→排卵。一般正常青春期发育是先出现乳腺发育,1.5~3年后才出现月经初潮。MAS患者的性早熟多在6岁前开始,平均发育年龄为3岁(最早为出生后1个月)。与正常的青春期发育并不相同,女性MAS患者的月经来潮是性早熟的首要症状,继而是乳腺发育(此与正常青春期发育不同),或者乳腺发育初现至月经初潮的时间少于18个月,而性毛生长因雄激素相对缺乏(肾上腺的性甾体类激素仍被抑制)而落后。血浆雌激素正常或升高。年幼患者血清LH和FSH对GnRH刺激无反应。女性性早熟与骨龄一致。当骨龄达到青春期年龄时,月经变得规律。在成人阶段,女性患者的青春发育期正常,并有生殖功能。
McCune-Albright综合征性发育特点

注:SCPP:继发性中枢性青春期发育提前(secondary central precocious puberty)又称为,混合性继发性中枢性青春期发育提前(combined secondary central precocious puberty,CSCPP)。
男性性早熟表现为精子生成、睾丸增大和第二性征提前发育。男性性早熟是由于Sertoli 细胞和Leydig细胞的功能过早启动所致。起病后,常先出现双侧性睾丸增大或单侧性巨睾,血清睾酮和抑制素B显著升高。随后出现阴茎增大和阴毛生长,青春期前患儿有时发生巨睾症而无阴毛、腋毛生长,阴茎未发育。LH对GnRH反应如正常儿童,FSH反应迟钝。这些改变提示,睾丸的Sertoli细胞处于高功能状态。睾丸组织学检查可见Sertoli细胞增生,精原细胞减少,间质含较多间叶细胞,曲细精管增大;但未见成熟的Leydig细胞。巨睾男性的雄激素分泌并无明显增多,可能与单纯性Sertoli细胞功能亢进或Gsα基因突变有关。睾丸微结石(testicular microlithiasis)为男性MAS的重要特征。
由于下丘脑长期受到性甾体类激素的作用,在MAS发病后某个阶段,可能提前发生继发性中枢性青春期发育提前(secondary central precocious puberty),并可能与原已存在的性发育一道,使青春期发育进一步提前,这种现象称为混合性继发性中枢性青春期发育提前(combined secondary central precocious puberty,CSCPP),是McCune-Albright综合征性发育的另一个重要特点。
Gsα突变引起多种内分泌代谢疾病或功能异常
肥胖
肥胖为Gsα基因突变的主要表型形式之一,其机制未明。Carel等用非放射性核素示踪稀释技术测定了6例Gsα突变伴PTH抵抗患者的脂肪细胞代谢情况,这些患者的基础甘油生成下降50%,脂肪细胞对肾上腺素的脂解反应下降67%,说明脂肪细胞对肾上腺素的脂解作用不敏感,从而导致肥胖,但在病态肥胖和轻度超重的儿童中,脂解作用受损的程度相近。
TSH相关性甲亢
是由于过多TSH(垂体无TSH瘤)刺激甲状腺所致,常出现甲状腺多发性结节。结节常为良性,放射性碘摄取率增高。甲状腺无淋巴细胞浸润,也无甲状腺兴奋性TSH受体自身抗体。其他的表现有囊肿或结节,虽然临床无甲状腺功能异常症状,但病理表现为弥漫性病变,并存在一定程度的功能自主性。治疗与一般的Graves病相同,可根据情况采用药物、131I或手术治疗,但从发病机制上考虑,可能以手术治疗的效果更佳。现已报道了两例MAS并甲状腺癌病例,但似乎两者无明确联系。
Cushing 综合征
MAS伴Cushing综合征不常见。儿童型Cushing综合征表现为生长迟滞,血ACTH降低,大剂量地塞米松不能抑制肾上腺的皮质醇和性甾体类激素的分泌。
肢端肥大症和高泌乳素血症
因性早熟或颅面骨质增生,MAS患者的GH分泌过多可被掩盖,不易获得早期诊断,常规测定血GH和IGF-1有助于较早发现并存的肢端肥大症。肢端肥大症症状与一般垂体GH瘤类似,血清GH增高且不受葡萄糖的抑制。颅骨的纤维发育不良也与肢端肥大症类似。因MAS病程进展缓慢,所以应定期扫描颅骨。MAS的肢端肥大往往与高泌乳素血症合并存在。
高磷酸尿和低磷血症性佝偻病或骨质软化
可与MAS合并存在,MAS患者尿cAMP增加,与肾小球cAMP的滤过增加有关。尿cAMP对外源性PTH反应迟钝。骨纤维增生不良症可单独出现或是MAS的特点之一。其病因为Gsα突变,病理学上可见大量异常增殖与分化的骨原细胞。但能表达FGF23的细胞除骨原细胞外,还包括成纤维细胞、骨细胞和成骨细胞。据报道,约一半的MAS患者伴有磷利尿和骨质软化症,血FGF23明显升高。有资料表明,FGF23分泌过多和VD代谢异常可能是导致高磷酸尿和低磷血症佝偻病或骨质软化的重要病因,但更可能的直接原因是肾小管上皮细胞的Gsα活化性突变。一般可用VD和口服磷酸盐治疗,但多数对治疗有抵抗。
假性甲旁减
Bastide-Eizaguirre等报道1例男性MAS伴有Ⅰa型假性甲旁减(PTH不敏感),而其母亲虽有AHO的临床表现,但无PTH抵抗(假性甲旁减),病因都是Gsα基因的第10号外显子的点突变(R265H)。
其他表现
肝脏异常包括严重的新生儿黄疸,肝酶活性增加。肝活检时发现胆汁淤积和胆管异常表现,有时可为MAS的首发症状,应注意与其他原因所致的先天性淤胆综合征鉴别。心脏的异常包括心脏扩大、心肌肥厚、持续心动过速和猝死。限制性肺疾患、动静脉分流或原发性心脏异常和心脏传导障碍的病因还不清楚。
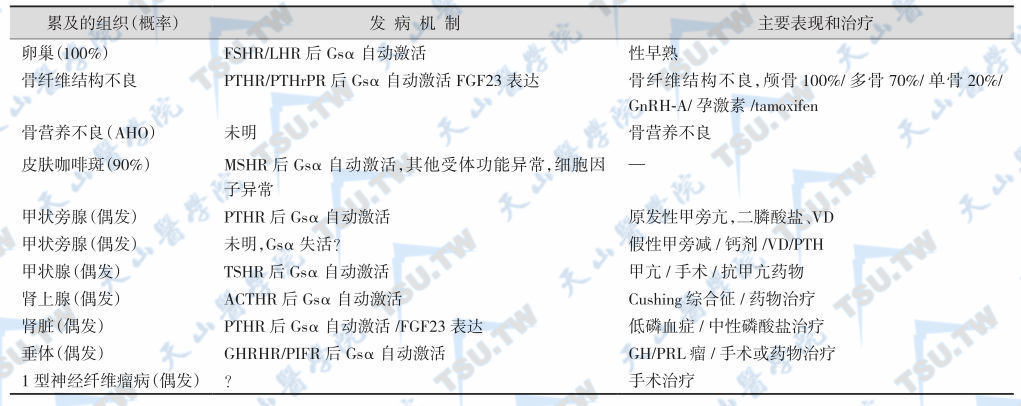
综上所述,McCune-Albright综合征受累组织的发病机制与表现见下表。
McCune-Albright综合征受累组织