
世界卫生组织(WHO)定义的骨质疏松症(osteoporosis,OP)是以骨量减少、骨组织微结构(microarchitecture)破坏、骨脆性增加和易于骨折为特征的代谢性骨病。2001年,美国国立卫生研究院(NIH)定义的骨质疏松症是以骨强度下降、骨折风险增加为特征的骨骼系统疾病。NIH的骨质疏松症定义强调了骨强度(bone strength),骨强度由骨矿密度和骨质量两个主要参数决定。但是,该定义仍然没有涉及骨微结构破坏的病理特征。从病理角度看,虽然骨质疏松和骨质软化(osteomalacia)都存在骨量减少、骨折风险增加和骨微结构破坏,但骨质软化的矿物质/骨基质比例下降,而骨质疏松不伴骨矿物质与骨基质比例的明显改变。
骨质疏松症可分为原发性(primary)和继发性(secondary)两型,前者又可分为绝经后骨质疏松症(postmenopausal osteoporosis,PMOP,Ⅰ型骨质疏松症)和老年性骨质疏松症(senile osteoporosis,SOP,Ⅱ型骨质疏松症,一般指70岁以上)两种。也有人将上述的Ⅰ型和Ⅱ型骨质疏松症统称为退行性骨质疏松症(degenerative osteoporosis)。特发性青少年低骨量/骨质疏松症(juvenile idiopathic osteopenia/osteoporosis,JIO)是原发性骨质疏松中的特殊类型,特指绝经前女性或年轻男性不明原因的低骨量(osteopenia)或骨质疏松。继发性骨质疏松症是指可以找到明确病因的一类骨质疏松,临床上以内分泌代谢病、结缔组织病、肾脏疾病、消化道疾病和药物所致者多见。但在临床上,原发性骨质疏松症患者往往合并有继发性骨质疏松,或继发性骨质疏松症患者涉及多种风险因素。本节重点介绍PMOP。
骨质疏松症是人类第6大常见慢性病。根据国际骨质疏松联盟(IOF)的统计,目前全球有2亿骨质疏松患者;美国、欧洲和日本的患病人数约7500万,我国7000万~8000万。在美国、英国和瑞士,骨质疏松占老年人口的60%,PMOP骨折率约为正常人的4倍。骨质疏松对患者、家庭和社会造成沉重的经济负担。据统计,中国目前有老龄人口(年龄>50岁)3.5亿,其中合并髋骨骨折(hip fracture)者约660万,占1.9%。至2025年,骨质疏松的流行将移至中东、亚洲、拉美和非洲,骨质疏松与其所致的骨折人数将成倍增加。
骨质疏松是中老年人群的常见病
骨质疏松的发生率随着年龄增长而迅速升高,在女性绝经后早期,骨丢失达3%~5%/年,至80岁时,髋部Ward三角骨丢失达60%。全世界每年的骨质疏松性骨折(osteoporotic fractures)患者约150万。在英国,骨质疏松所致髋部骨折占骨科患者的20%,其中80%是老年妇女。由于生活不能自理,活动受限,加上肺部感染、营养不良和加速的失用性骨质疏松,患者多在数月至2年内死亡。脊椎压缩性骨折的致残致死率也很高,5年存活率约2/3;年龄越大,死亡率越高。骨质疏松性骨折已成为目前很多国家的严重社会问题和医疗问题。50年后,全球的骨质疏松性骨折患病人数平均增加3~4倍。
美国白人妇女50岁以上的脊椎压缩性骨折发生率为每年18‰,显著高于髋部骨折发生率(每年6.2‰),女性脊椎压缩性骨折约为男性的2倍,明显高于男性,农村高于城市。前臂远端骨折的主要诱因是摔倒。骨质疏松还可导致肱骨、骨盆、尺骨骨折。人群中,这些部位的骨折发生率亦随年龄增加而升高,女性约占3/4。北京等地区基于影像学的流行病学调查显示,50岁以上妇女脊椎骨折患病率为15%,相当于每7名50岁以上妇女中就有1人发生过脊椎骨折。近年来,我国髋部骨折的发生率也有明显上升趋势。
随着人类平均寿命的延长,骨质疏松已成为中老年人群的常见疾病之一,其发病率呈上升趋势,预计到2050年将增加到2亿,那时全世界一半以上的骨质疏松性骨折将发生在亚洲,绝大部分患者在我国。
骨质疏松性骨折的常见部位是脊椎、髋部和前臂远端。骨质疏松性骨折的危害大,导致病残率和死亡率增加。如发生髋部骨折后1年之内,死于各种并发症者达20%,而存活者中约50%致残,生活不能自理,生命质量明显下降。而且,骨质疏松和骨质疏松性骨折的治疗和护理需要投入巨大的人力和物力,费用高昂。值得强调的是,骨质疏松性骨折是可防可治的,尽早预防可以避免骨质疏松及其骨折。即使发生过骨折,只要采用合理治疗仍可有效降低再次骨折的风险。因此,普及骨质疏松知识,作到早期诊断、及时预测骨折风险并采用规范的防治措施是十分重要的。
可变危险因素和不可变危险因素引起骨丢失
PMOP的病因主要是雌激素缺乏,但发病机制尚未阐明。导致PMOP的危险因素(risk factors)很多,这些因素作用于成骨和破骨的某些阶段,最终使骨量丢失。显然,单纯的雌激素缺乏只是其中的重要原因而非全部。骨质疏松的危险因素包括不可控制因素和可控制因素两个方面。不可控制因素主要包括人种(白种人和黄种人患骨质疏松的危险高于黑人)、老龄、女性绝经、骨折家族史(重点是父母65岁以前的髋部骨折史)等;可控制因素包括低体重、性激素缺乏、吸烟、过度饮酒、体力活动不足、钙和维生素D(VD)缺乏及药物等。
遗传因素
原发性骨质疏松症的遗传易感性较强,骨质疏松与脆性骨折的风险基因及其效应可分为以下几类:
- 效应大的低变异频率基因(如LRP5、SOST、COL1A1、COL1A2、LEPRE、CRTAP、PpiB等);
- 效应大的高变异频率基因(似乎很罕见);
- 效应小的高变异频率基因;
- 效应小的低变异频率基因(如TNFRSF11A、TNFRSF11B、ESR1、SP7、LRP4、LRP5、THFSF11、SOST、MRRK3、ZBTR40等);
- 效应中等的高变异频率基因(意义大,应列为重点研究对象,但鉴定极为困难)。
骨吸收增强
一、妊娠期
骨吸收增强导致骨小梁变细、变薄甚至断裂,骨微结构有明显变化。妊娠期妇女对钙、磷的需要量较非妊娠妇女增加1倍,尤其是妊娠中期以后,胎儿发育需要的钙量大,随着孕周延续,母体缺钙易出现腓肠肌痉挛、腰腿痛等表现。虽然正常妊娠对母亲的骨代谢有明显影响,但一般通过代偿不至于发生严重骨丢失;但多次妊娠加上蛋白质、热能、钙和VD等的摄入不足或其他一些原因,可成为PMOP的高危对象。
正常哺乳妇女每天从乳汁丢失200~250mg元素钙,如为双胞胎或同时哺乳两个以上婴儿,每天从乳汁丢失的元素钙可达600mg。显然,适当补充钙的摄入量有充分理由。但补钙不能完全纠正负钙平衡和骨盐丢失。如妊娠中后期出现骨痛,DXA检查发现BMD下降,应视为异常,并需长期追踪。如果哺乳期间使用肝素则更易发生PMOP。
二、哺乳期
催产素刺激成骨细胞分化、骨矿化和破骨细胞形成,因此催产素是一种促进骨形成激素,哺乳期催产素升高在预防过度骨丢失与促进骨形成方面起了重要作用。根据以上分析,人们提出了垂体-骨轴(pituitary-bone axis)的概念。出生后哺乳需再动用约80g骨钙,因此骨吸收明显增强。但此后的骨形成加速可使骨量基本恢复正常。骨形成不足引起妊娠相关性和哺乳相关性骨质疏松(pregnancyassociated and lactation-associated osteoporosis)。
三、雌激素缺乏
性腺甾体类激素(gonadal steroid hormones)为青春期骨骼突发生长(growth spurt)的始动因子,生长发育延迟可引起PMOP。雌激素和雄激素(androgens)对成骨细胞和骨细胞的作用主要来源于“核受体”功能,但还存在雌激素膜受体,并与细胞外信号调节激酶的信号转导、MAPK及Src/Shc途径(位于胞质小泡中)有关。雌激素缺乏使非核受体作用减弱,破骨细胞和成骨细胞生成均增加,骨重建速率升高。加上成骨细胞和骨细胞凋亡,导致骨形成和骨吸收失平衡,骨吸收多于骨形成。
雌激素靶细胞表达4种雌激素受体(estrogen receptors,ER),分别称为ERα66、ERα46、ERα36和ERβ;其中ERα66 与ERα46为经典的核受体,而ERα36为膜受体 (图6-37-40)。既往认为,G蛋白耦联受体30(G protein coupled receptor 30,GPR30)介导雌激素的非经典膜效应;研究证明,GPR30通过结合ERα36基因启动子区的激活蛋白-1(AP-1)位点而促进ER-α36表达;GPR30表达被抑制后,ERα36表达也减弱,GPR30本身并不介导雌激素膜效应,其雌激素膜效应是通过ERα36实现的。我们的研究发现,雌激素膜型受体ERα36在绝经后妇女骨代谢中发挥了关键性调控作用。绝经后,ERα36在骨细胞高表达,发挥骨保护作用。如果缺乏这一反应,即可引起PMOP。
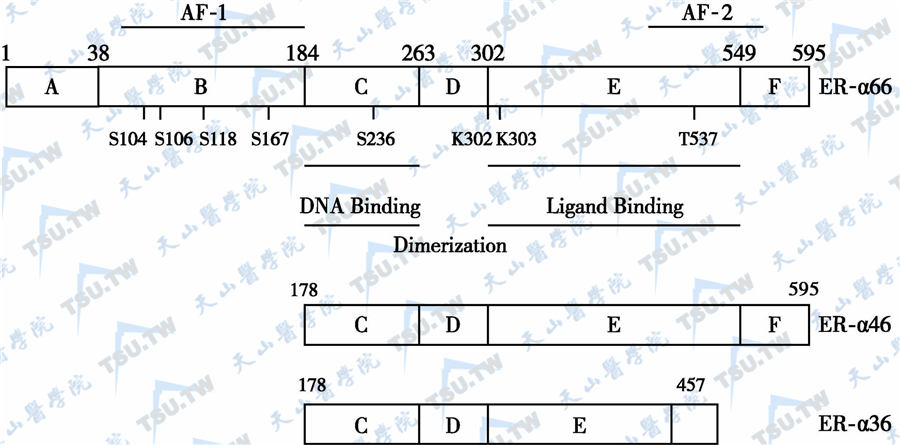
雌激素受体亚型的结构示意图
注:ER-α36缺乏两个转录功能区AF-1和AF-2,从而不介导雌激素经典效应(核效应)。ER-α36保留了完整的DNA结合区和部分配体结合区,主要在细胞膜和细胞质表达,介导雌激素非经典快速膜效应。既往认为,G蛋白偶联受体30(G protein coupled receptor 30,GPR30)介导雌激素的非经典膜效应。最新的研究结果证明,GPR30通过结合ER-α36基因启动子区的激活蛋白-1(AP-1),促进ER-α36表达;GPR30表达被抑制后,ER-α36的表达也减弱;GPR30本身不介导雌激素膜效应,GPR30介导的雌激素膜效应是借助ER-α36实现的。基因敲除鼠试验证明:ER-α介导雌激素对骨和子宫的作用, ER-β则否。
IC162和SNG8006是植物异黄酮的两种衍生物,它们的作用依赖于ERα36,促进成骨细胞增殖和分化,抑制成骨细胞凋亡,促进破骨细胞凋亡。研究发现,IC162和SNG8006能防止去卵巢大鼠骨丢失,且无子宫增重作用(正常子宫无ERα36表达)。在低水平E2情况下(如绝经后),机体的雌激素靶细胞依靠ERα36高表达而部分代偿雌激素缺乏。因而,通过刺激ERα36表达可达到补充雌激素的目的而不出现经典雌激素的不良反应。
绝经是雌/孕激素同时缺乏的状态,孕激素受体(progesterone receptor,PR)有A、B两种异构体,两者的基因相同而转录所需要的启动子不同,但两种启动子均可被雌激素诱导活化,一般B型(PR-B)异构体的转录活性强于PR-A,而PR-A可下调PR-B和其他甾体类激素受体(尤其是雌激素受体)的转录活性。在许多情况下,可能存在雌激素/孕激素受体对话(cross-talk)现象。因此,雌激素对骨代谢的一些作用可能是通过PR介导的。在临床上,存在卵巢黄体期功能缺陷者均易发生骨质疏松,而使用孕激素后骨量增加。
围绝经期妇女因卵巢功能减退,抑制素B下降使FSH分泌增高,骨组织的活化素activin/骨形态生成蛋白(BMP)调节失常,破骨细胞活性增强,骨转换水平升高。至绝经后,抑制素A和雌二醇减少,FSH分泌、破骨细胞活性和骨转换水平进一步升高,骨丢失逐年加重。
四、去氢异雄酮和雄烯二酮不足
无论是雄性还是雌性动物,雄激素和雌激素的作用都是不可缺少的。在骨细胞上,雄激素受体和P450芳香化酶为雄激素调节骨代谢提供了两种途径:①直接通过雄激素受体发挥作用;②在骨微环境中将雄激素芳香化为雌激素而起作用。雄激素也可通过调节骨微环境中的细胞因子、生长因子(包括IL-6、IGFs、TGFβ和FGF等)的产生来调控骨代谢。女性的雄激素来源于卵巢、肾上腺和脂肪组织;卵巢主要生成睾酮和二氢睾酮,肾上腺生成和分泌去氢异雄酮及其硫酸盐和雄烯二酮。绝经后妇女的血睾酮及其他雄性甾体类激素均明显下降,血去氢异雄酮硫酸盐与腰椎、股骨颈和桡骨BMD呈正相关,而选择性雄激素受体调节剂(selective androgen receptor modulator,SARM)对男性骨质疏松有治疗作用。
五、VD缺乏/不足
慢性VD不足是骨质疏松的常见病因,而重度VD缺乏导致骨质软化(osteomalacia)/佝偻病(rckets)。1,25-(OH)2D可加速小肠绒毛细胞成熟,促进钙结合蛋白(calcium-binding protein,CaBP)生成,增加肠钙吸收。VD对骨组织具有两重性,生理量1,25-(OH)2D刺激成骨细胞活性,促进骨形成;但大剂量可激活破骨细胞,增强破骨细胞的骨吸收作用。VD缺乏导致继发性甲旁亢和骨质软化症,轻度不足则表现为骨质疏松症。成骨细胞表达VD受体(VD受体,VDR),而VD可调节成骨细胞多种靶基因表达。另一方面,24-羟化后的代谢产物24,25-(OH)2D曾被认为是VD的降解产物。但近年发现,这种VD衍生物仍有骨代谢调节作用,而且还可以促进骨折愈合。
维生素D是经典的内分泌——旁分泌激素,钙代谢和其对机体的影响与VD密切相关。钙和VD缺乏对细胞增殖-分化的影响途径主要在成骨细胞。成骨细胞增殖分化低下使骨形成减少,骨量不足。全美第三次健康和营养调查的资料显示,老年人血清25-(OH)D明显低于正常,并随年龄增长而进一步下降,85岁时仅相当于10岁前儿童水平;与此同时,髋部BMD降至峰值骨量的50%。
钙和VD缺乏对细胞增殖和分化影响的次要途径在成克隆细胞(colonocytes)。克隆细胞增殖亢进促进肿瘤形成,分化障碍则引起免疫功能紊乱、慢性应激和代谢异常。钙与VD慢性缺乏/不足状态对人类健康的影响远远超过人们已有的认识。近年,人们提出了钙/VD缺乏相关性疾病的概念,其涉及的范围相当广泛,并将其可信度分为4个等级:
- A级证据(有力证据,资料来源于多个大型流行病学/RCT干预/试验研究):VD缺乏相关性疾病有骨质疏松症、直肠-结肠癌、乳腺癌,钙缺乏相关性疾病有骨质疏松症、直肠-结肠癌、乳腺癌;
- B级证据(充分证据,资料来源于3个以上的良好观察或干预研究):VD缺乏相关性疾病有肿瘤(肾、前列腺、子宫、卵巢、食管-胃、胰腺、膀胱、淋巴瘤)、心血管病、神经肌肉疾病、1型糖尿病、结核病、牙龈炎、牙周病,钙缺乏相关性疾病有心血管病、高血压、牙周病、神经肌肉疾病、肾癌;
- C级证据(一般证据,资料来源于观察性研究):VD缺乏相关性疾病有高血压、代谢综合征、T2DM,钙缺乏相关性疾病有代谢综合征和T2DM;
- D级证据(间接证据,资料来源于相关疾病的动物模型研究):钙与VD缺乏相关性疾病有炎性肠病和多发性硬化症等。
六、不良生活方式
吸烟、酗酒、高蛋白、高盐饮食、VD摄入不足和光照减少等均为骨质疏松的易患因素。吸烟通过干扰骨骼肌功能而引起骨丢失。烟草中的苯并芘(benzoapyrene,BAP)和7,12-二甲基二苯蒽(7,12-dimethyl benzanthracene,DMBA)均为多环芳香羟化合物(polycyclic aromatic hydrocarbons,PAH)。BAP和DMBA存在于污染的大气、汽车尾气和液化石油气中,长期接触者易发生骨质疏松。慢性酒精中毒伴有严重骨丢失。除肝功能不全、脂代谢紊乱和蛋白质缺乏等因素外,乙醇对骨组织也有某种直接作用。
七、肥胖
肥胖与骨代谢的关系复杂,骨质疏松和肥胖均为发病率剧增的常见病,均有明显的遗传背景,而脂肪细胞和骨细胞来源于共同的干细胞。增龄性肥胖后,骨髓、脂肪细胞增多,破骨细胞活性增强,而成骨细胞功能减退。另一方面,糖尿病、糖皮质激素或制动(immobilization)引起的骨质疏松伴有骨髓脂肪沉积(bone marrow lipidosis)。研究提示,骨髓脂肪沉积与骨质疏松相关。间充质干细胞可分化为成骨细胞、内皮细胞、成纤维细胞、脂肪细胞或软骨细胞,脂肪细胞、成骨细胞、破骨细胞均调节骨代谢,脂肪细胞与成骨细胞的分化存在相互抑制现象。肥胖者脂肪细胞分泌TNF-α、IL-1β、IL-6、leptin等,抑制成骨细胞分化,促进破骨细胞生成;内脏脂肪(visceral adipose tissue,VAT)/皮下脂肪(sc adipose tissue,SAT)比值与腰椎及全身BMC(WB-BMC)、BMD负相关,肥胖T2DM诊断时骨折风险增高约30%,骨折风险随病期延长而升高,30年后达50%。因此体脂比率升高是骨丢失的风险因素,而减肥能降低骨折风险。肥胖不是骨健康的保护因素,一些轻度肥胖者BMD较高的原因可能是其他营养因素引起的。
骨形成不足
骨形成主要由成骨细胞介导。在成骨过程中,向基质分泌胶原蛋白和其他基质物质,为矿物质的沉积提供纤维网架,然后类骨质被矿化为正常骨组织。人类在30岁左右达到一生的骨量最高值(骨峰值,peak bone mass,PBM)。青春发育期是人体骨量增加的最快时期,如因各种原因导致骨骼发育和成熟障碍致PBM降低,成年后发生骨质疏松的可能性增加,发病年龄提前。PBM越高,发生骨质疏松的可能性越小或发生的时间越晚。因此,影响人体骨量的另一因素是增龄性骨丢失前的PBM。达到PBM年龄以后,骨质疏松主要取决于骨丢失的量和速度。PBM主要由遗传素质决定,但营养、生活方式和全身性疾病等对PBM也有明显影响。
一、峰值骨量较低
PBM是遗传因素和环境因素共同作用的结果,一般自幼体健、具有健康素质的个体和青春期发育正常者PBM较高。出生时体重、生活习惯、健康状态、体力活动为主要的影响因素,而男、女性的PBM影响因素又有所不同。后天性不利于获得较高PBM的因素多是可以预防的。例如,保证钙的摄入量和加强体育运动有助于获得更高的PBM。
1)决定PBM和BMD的遗传因素:
人群中的峰值骨量和成年后骨丢失速度不同是支持遗传因素影响骨质疏松发病的有力证据;一般认为,遗传因素对峰值骨量的影响最大,而成年后的骨丢失速度主要由环境因素决定,但所涉及的基因数目、染色体定位、影响程度及相互作用方式尚未确定。胎儿的生长发育受遗传因素和环境因素影响,遗传素质、母亲吸烟和体力活动均对胎儿的骨发育有影响,其中新生儿低体重与BMD的关系最密切。
BMD仅是决定骨生物质量的一个方面,骨基质的质和量对骨质疏松及其骨折的发生也起重要作用。腕部骨折很难用全身或局部BMD下降来解释,Ⅰ型胶原的α-1基因(COL1A1)的第1号内含子Sp1多态性与腕部骨折有关。COL1A1基因多态性可能有较大的种族差异,该基因对BMD和骨质疏松的影响尚需在不同人群中进一步研究。
2)决定股骨颈几何形态的遗传因素:
由遗传因素决定的股骨颈几何形状(geometry)和生物质量(bioquality)存在种族差异,股骨颈骨折与其他骨折不同,在同等外力作用下,股骨颈是否骨折与其长度、宽度、直径、Ward三角形状等有关。因而,预测股骨颈骨折危险性时,除考虑BMD外,还应将该部位的几何形态参数作为预测因素。
3)钙摄入不足:
钙是骨矿物质中主要成分,钙摄入不足必然影响骨矿化。在骨的生长发育期和钙需要量增加(妊娠、哺乳等)时,摄入钙不足将影响骨形成和PBM。增加钙摄入量有助于防治骨质疏松,降低骨折风险。
二、肌量减少/体重过低
瘦体重(肌容量)是骨保护因素。个体在达到PBM后,一生中要减少20%~30%的骨骼肌组织,这一现象称为肌量减少(sarcopenia)。随着增龄而减少骨骼肌量的原因很多。肌量下降使活动能力降低,而体力活动下降、食欲不振和平衡能力差又进一步加重肌肉消耗,形成肌量减少和骨丢失之间的恶性循环。
三、体力活动不足
成骨细胞和骨细胞具有接受应力、负重等力学机械刺激的接受体(acceptor),足够的体力活动有助于提高PBM和维持骨量,故成年后的体力活动是刺激成骨细胞的基本方式,活动过少易于发生骨质疏松。此外,由于主动或被动原因使机体制动,骨骼失去机械应力刺激,成骨细胞活性被抑制,而破骨细胞活性增强,导致“失用性骨质疏松(disuse osteoporosis)”。这种骨质疏松的特点是发生于经常负重的骨骼部位。长期卧床(long-term bed)和失重也常导致骨质疏松。
四、药物与放疗
可导致骨质疏松的药物很多,最常见的是糖皮质激素、化疗药物、抗凝剂和抗惊厥药,各种药物引起骨质疏松的作用机制不同。放射性骨坏死(osteoradionecrosis)是骨组织放射治疗中的严重并发症,表现为骨愈合能力衰竭和自发性骨坏死。组织学上,开始表现为骨形成缺陷伴破骨性骨溶解,继而出现骨纤维化。
内分泌激素分泌紊乱引起骨丢失
内分泌激素分泌紊乱导致绝经后妇女骨丢失见于雌激素、VD和去氢异雄酮/雄烯二酮缺乏,而PTH和FSH分泌增多是引起骨丢失的重要原因。
雌激素与VD和去氢异雄酮/雄烯二酮缺乏
雌激素、VD和去氢异雄酮/雄烯二酮是促进骨形成的必需激素,增龄引起VD缺乏和去氢异雄酮/雄烯二酮不足,因卵巢功能衰竭出现雌激素缺乏,进而引起骨形成不足与骨吸收增强。
FSH升高
绝经期FSH升高与骨丢失增多相关,绝经后5年内,骨丢失量占绝经后骨丢失总量的50%以上。FSH通过Gi2α-耦联的FSH受体直接刺激破骨细胞形成和骨吸收,促进受体下游RANKL激酶磷酸化,抑制NF-κB与IκBα。以上3条途径均诱导骨吸收。FSH也刺激骨髓巨噬细胞释放TNF-α,导致骨丢失。
PTH增多
少量的PTH刺激骨形成。绝经后,部分患者血PTH和血钙轻度升高(游离钙升高为主),骨吸收指标明显升高,出现原发性甲旁亢样表现,符合“绝经后原发性甲旁亢”(postmenopausal primary hyperparathyroidism,PPHPT)的特点。一般认为,PPHPT是PMOP中的特殊亚型,但也有人认为是独立于PMOP的原发性甲旁亢类型,因为PPHPT与甲状旁腺主细胞增生所致的原发性甲旁亢并无本质区别。
局部调节网络功能紊乱引起骨丢失
在大多数PMOP患者中,调节钙、磷代谢的内分泌激素,如PTH、降钙素、VD和FGF23(排磷素,phosphorin)均无显著变化,所以骨丢失不是(或不主要是)这些内分泌激素调节紊乱引起的。IL-6为一种多功能细胞因子,可促进破骨细胞的分化和活性,刺激骨吸收。单核细胞和巨噬细胞分泌IL-6,诱导骨原细胞分化为破骨细胞。TGF-β和TNF促进骨吸收,加速骨丢失。另一方面,随着年龄增加,成骨细胞骨保护素(OPG)表达下降,骨丢失加速。局部调节网络功能紊乱导致骨丢失的其他依据有:①钙摄入不足、阳光照射少和消化功能减退引起血钙下降,导致轻度继发性甲旁亢;②细胞因子使骨组织对PTH的反应敏感性降低;③GH脉冲性分泌消失,血清IGF-1下降。
