
遗传性VD抵抗性佝偻病综合征分为两型。Ⅰ型维生素D抵抗性佝偻病(vitamin D resistant rickets type Ⅰ,VDRR1)是由于25-(OH)D的1α-羟化酶缺陷(vitamin D 1αhydroxylase deficiency)引起的骨骼矿化障碍性疾病,以前称为Ⅰ型维生素D依赖性佝偻病(vitamin D dependent rickets typeⅠ,VDDR-Ⅰ;OMIM 264700)或假性维生素D缺乏性佝偻病(pseudovitamin D deficiency rickets,PDDR)。从VD的作用机制缺陷方面看,患者的VD靶组织对25-(OH)D是抵抗的,但对1,25-(OH)2D是敏感的。为了与Ⅱ型维生素D抵抗性佝偻病的名称对应,并与VD缺乏引起的佝偻病/骨质软化区别,称之为Ⅰ型维生素D抵抗性佝偻病可能更贴切。VDRR1是一种常染色体隐性遗传性疾病,属于遗传性佝偻病(hereditary rickets)中的一种。遗传性佝偻病少见,发病率低,但临床漏诊可能不少。据丹麦的研究报道,遗传性佝偻病的总体发病率为5.2/10万,低磷血症性佝偻病为4.8/10万,而VDDR-Ⅰ为0.4/10万。
目前已经发现了36个突变位点,其中958delG(外显子8插入7个外来核苷酸)和IVS3+1G>A两种突变较常见。临床表现以肌张力低下,肌肉无力、生长发育障碍、低钙血症、低(正常)磷血症、高PTH血症、高ALP血症为特征,血清25-(OH)D可正常、升高或降低,1,25-(OH)2D显著降低。影像检查可见骨骼呈佝偻病改变。
VDDR1的临床表现与维生素D缺乏性佝偻病相似
VDDR1的临床表现与维生素D缺乏性佝偻病基本相同,但起病更早(一般在2岁前发病),症状更重,进展更快,佝偻病的生化改变和影响表现更突出。儿童期发病的佝偻病应重点询问遗传家族史,如果血清钙磷均降低,血清25-(OH)D正常或升高,而血清1,25(OH)2D显著下降,基本可以确立VDDR1诊断。VDDR1患者容易并发多发性硬化症(multiple sclerosis,MS),其原因未明。流行病学研究发现,多发性硬化症的发病与维生素D缺乏相关,动物实验证实,1,25-(OH)2D具有免疫抑制作用。Torkildsen等报道了3例VDDR1患者均合并有多发性硬化症,表现为脑皮质萎缩,MRI T2高信号及膑骶体萎缩(图3-12-28)。此外,VDDR1偶尔合并苯丙酮尿症(phenylketonuria,PKU),因为PAH和 CYP27B1基因均位于12号染色体的长臂,缺失或插入突变可引起两种基因同时失活。
Ⅰ型维生素D依赖性佝偻病(1α-羟化酶缺陷症)和Ⅱ型维生素D抵抗性佝偻病(维生素D受体缺陷症)均为遗传性佝偻病,两者的临床表现相似,应注意鉴别。
Ⅱ型遗传性VD抵抗性佝偻病对1,25-(OH)2D作用抵抗
Ⅱ型遗传性VD抵抗性佝偻病的缺陷在VDR对1,25-(OH)2D抵抗,又称遗传性1,25-(OH)2D抵抗性佝偻病(HVDRR)、Ⅱ型VD抵抗性佝偻病(vitamin D resistant rickets type Ⅱ)、假性VD缺乏性佝偻病Ⅱ型(pseudovitamin D deficiency rickets Ⅱ,PDDR-Ⅱ)、钙三醇抵抗性佝偻病(calcitriol-resistant rickets)、VD抵抗性佝偻病(vitamin D-resistant rickets)、遗传性低血钙性VD抵抗性佝偻病(hereditary hypocalcemic vitamin D-resistant rickets)等。因该综合征是由于对VD遗传性抵抗所致,故文献多认为该综合征应命名为遗传性VD抵抗性佝偻病(hereditary vitamin D-resistant rickets,HVDRR)。
HVDRR于1978年由Brooks等首先报道,其特点为:①有佝偻病或骨质软化的临床表现,常于出生后不久发病;患者有骨痛、肌无力、肌张力下降或低血钙性手足搐搦;患儿生长发育延迟,牙齿发育停滞等。可因并发肺炎等疾病而死亡。有些患儿头发稀少或全秃(包括眉毛缺如)。②血清中1,25-(OH)2D升高。③低钙、低磷血症,伴血清碱性磷酸和PTH升高,血清25-(OH)D正常,但1,25-(OH)2D明显升高,24,25-(OH)2D正常或下降,这些指标在使用大剂量VD后不能得到改善。④继发性甲旁亢。⑤全秃。⑥有家族发病倾向,呈常染色体隐性遗传。本病罕见,迄今世界文献中已报告的病例约数十例。
本病为遗传性疾病,但遗传缺陷存在不均一性。遗传方式为常染色体隐性遗传,发病呈家族性,男女发病几率相等,且常一个家庭有几个患儿。患者父母近亲结婚者多,无临床症状且骨骼发育正常,多为表型正常的杂合子,患者则为纯合子。其病因为VDR基因突变,导致VDR功能异常。
肾近曲小管细胞的刷状缘膜(brush border membrane,BBM)表达Na/Pi酮转运体(Na/Pi cotransporters)Na/Pi-Ⅱa和Na/Pi-Ⅱc,调节磷的重吸收,Na/Pi-Ⅱc突变引起遗传性低磷血症性佝偻病伴高尿钙症(hereditary hypophosphatemic rickets with hypercalciuria,HHRH)。
HVDRR表现为VDR数目减少/亲和力降低/受体蛋白结合减少
Ritchie等报告无亲缘关系的4例患者,在C末端外显子7的第970位胞嘧啶突变为腺嘌呤(TAC→TAA),编码的VDR氨基酸密码子(TAC)变为终止密码子,VDR基因提前终止转录,使VDR蛋白被截去292个氨基酸,其中包括VDR的配体结合区(ligand-binding domain,LBD)在内,使VDR分子量减小(正常为50kD),也有被截去291和362个氨基酸者,结果VDR不与1,25-(OH)2D结合,称之为VDR结合阴性型。VDR的LBD区的常见突变类型见图6-37-60和图6-37-61。患者为纯合子,其父母为杂合子,如R30stop突变型VDR缺失398个氨基酸残基(包括锌指结构的大部分及全部激素结合结构域),这种病例的病情往往更为严重。Malloy报道一例亚洲患者,临床表现典型,于第6外显子发现错义突变(T→G),编码的蛋白251位氨基酸苯丙氨酸变为半胱氨酸,使得VDR数量及亲和力均下降,干扰RXRα异二聚体形成而导致发病。另一例HVDRR患者亦具备该病各临床特点,且严重体秃,基因测序示第8外显子点突变(C→T),使得蛋白产物317位氨基酸密码子变为终止密码子,导致VDR的LBD缺失110个氨基酸,VDR不能与1,25-(OH)2D结合而发病。
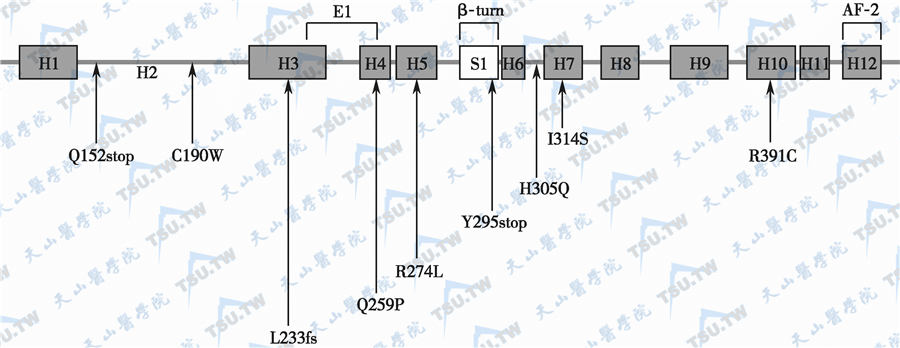
VDR蛋白的配体结合(LBD)区和该区的部分突变位点
注:LBD区突变导致配体结合阴性反应。图中的H1~H12代表VDR蛋白配体结合区的α-螺旋;S1代表β-折叠区;fs代表读码框架(framesheft)
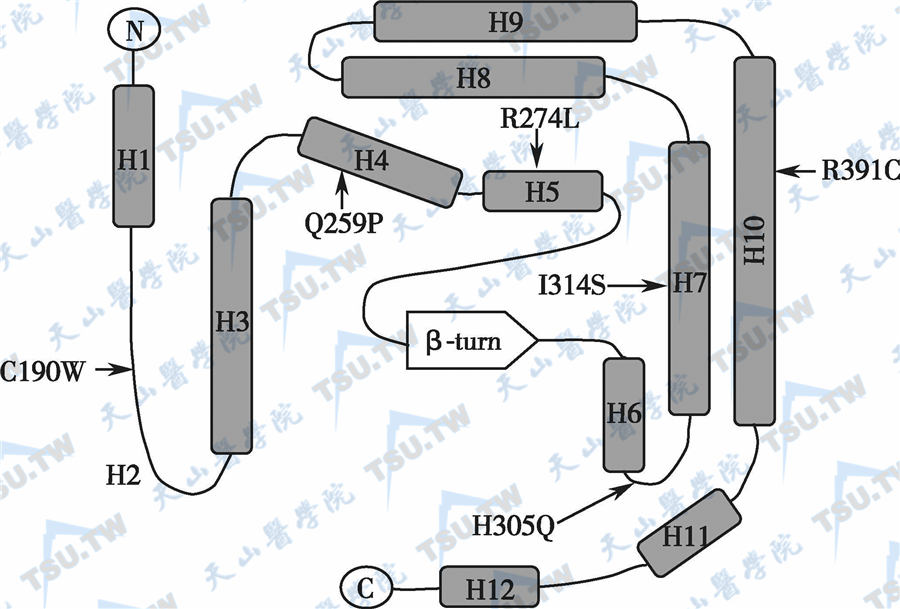
VDR的配体结合区及其突变位点
注:图中箭头所示为点突变位点,H1~H12代表α-螺旋区,β-turn为β折叠处。图中所标出的突变均导致HVDRR
VDR结合量与亲和力正常而与DNA反应元件结合缺陷
DNA的反应元件含有两个锌指,锌指中如有突变,则引起锌指功能异常。故此类突变多见于外显子2和3。文献中已报告的锌指区点突变类型有:
- VDR基因第一锌指顶端突变(如GGC→GAC突变导致在VDR中的甘氨酸被门冬氨酸取代)。
- 两锌指间的内含子突变(如CGG→CAG突变使VDR中的精氨酸被谷氨酸取代)。
- 第二锌指顶端突变(如CGA→CAA突变导致VDR中的一个精氨酸被谷氨酸取代)。
- 第二锌指的底部突变(如CAC→CGG或点突变导致与上述相似的氨基酸突变)。
- VDR基因在外显子3突变(如第146位鸟嘌呤突变为腺嘌呤,使VDR的第47位的精氨酸被谷氨酰取代)。
前述VDR基因的突变也使VDR电荷发生变化,使VDR复合物不能进入细胞核与DNA结合,1,25-(OH)2D与DNA结合减少。Malloy报道1例HVDRR患者,临床表现典型,但无秃头,行序列分析后发现VDR基因第7外显子上点突变(T→C),相应蛋白产物的268位氨基酸异亮氨酸由苏氨酸占据,导致与1,25-(OH)2D亲和力较野生基因型下降80%~90%。其报道的另一HVDRR患者,亦无秃头表现,于第4外显子发现5bp缺失/8bp插入,则LBD缺失2个氨基酸(H141/T142)/插入3个氨基酸(L141、W142 和A143),使突变型VDR的活性较野生型减低1000倍。
VDR核定位缺陷的患者的成纤维细胞可溶性提取物与1,25-(OH)2D有正常的或近乎正常的结合,但在完整细胞中未能检出1,25-(OH)2D定位于细胞核,如:①ATC→AGC (I314S)突变;②CGC→TGC(R391C)突变。推测这些患者存在有对核定位有重要作用的二聚体化有缺陷。
VDR结构和功能及结合反应正常而诱导生成减少
正常人皮肤成纤维细胞与1,25-(OH)2D同时温育8小时,可使加入媒液中的25-(OH)D转化为24,25-(OH)2D的量比未加1,25-(OH)2D的对照细胞的量增加20倍,有此缺陷的患者则明显减少[0.11~0.27pmol/(min·mg)蛋白对0.02pmol/ (min·mg)蛋白]。
BAG-1 P50、-P46、-P33、-P29为人体中的4种抗细胞凋亡蛋白的异构体。BAG-1P46与甾体类激素受体结合并调节其活性,BAG-1 P50可与VDR相互作用,或促进VDR与 DNA反应元件结合,继而抑制靶基因的转录,细胞生长加速,且阻滞了1,25-(OH)2D诱导的细胞增殖作用,使1,25-(OH)2D不能诱导VDR的活性,因此BAG-1 P50是一种VD信号转导途径的调节因子,如果表达过度可导致靶细胞对1,25-(OH)2D治疗的抵抗。一个经典的点突变即为VDR的LBD第420位谷氨酸由赖氨酸替代(E420K),与甾体激素受体共激活剂-1(SRC-1)及DRIP205结合功能受损从而导致本病的发生。
VD作用障碍导致肠钙磷吸收障碍与继发性甲旁亢
本病多见于1~2岁婴幼儿,亦有至年长或成年发病者。发病呈家族性或散发性。患者父母为病态基因携带者,近亲结婚者多,临床上可无任何异常(杂合子)。由于遗传缺陷的不均一性,临床表现除取决于VDR功能受损的严重程度外,与治疗早晚也有关。在正常情况下,约50%的肠吸收钙依赖于1,25-(OH)2D的促进作用,另50%以被动性吸收方式进入循环血液中。由于DVR突变,肠钙磷吸收困难,血钙降低,导致继发性甲旁亢,加重低磷血症。由于低血钙和低血磷,骨骼不能正常矿化,儿童则发生佝偻病,故早期常有骨痛。
低血钙症严重者甚至可有癫痫大发作样抽搐。体检时叩面征(Chvostek)和束臂征(Trausseau)阳性,易发生骨骼畸形(成人易发生骨折)。常见的骨骼畸形在下肢,如膝外翻、胫腓骨弯曲,其次为上肢和脊柱。患儿骨骼生长延迟,某些有严重龋齿或牙发育不全。患者身材矮小,常低于同龄正常儿童身高的两个标准差。随着年龄的增大,部分患儿骨痛缓解,骨折自发性愈合,癫痫大发作和骨骼畸形发展也停止,这可能是由于随着年龄的增大,肾脏中的1α-羟化酶趋于成熟所致。此外,尚伴有继发性甲状旁腺功能亢进症。
毛囊VDR功能障碍引起毛发异常
患者在用大剂量1,25-(OH)2D治疗后,低钙血症及骨骼病变有好转,但全秃无缓解。一般认为,毛发缺失越严重,其对VD的抵抗也越明显,如新生儿有毛发缺失,尤其是全秃或体秃者要首先想到本征可能。近年来的散发病例报道患者具备各典型表现,而无全秃,毛发生长正常。
全身性毛发缺失(generalized atrichia)伴皮肤小疹是由于无毛基因(hairless gene,HR)突变所致的一种遗传性疾病,但有些患者的HR基因并无异常,而是VDR突变引起的。由于VDR基因突变引起的毛发缺失者,皮肤毛囊发育障碍,常伴毛囊囊肿,囊肿上皮含有角质素(keratin)-15和-17阳性细胞(来源于毛囊上皮干细胞)。VDR和HR的表达产物均为含有锌指结构的蛋白质,是调控出生后毛囊生长和代谢的重要因子,可能两者的作用途径相同。因此,HVDRR所伴有的毛发缺失和毛发发育障碍可能与HR基因的结构或功能异常有关。
血PTH升高伴骨骼普通性脱钙
- 血液生化:血钙低、ALP升高,骨源性ALP增高,血磷大多正常,也可有PTH升高,低磷血症(继发甲旁亢)。
- 尿液指标:尿钙排泄减少,cAMP增高(与PTH升高有关)。少数患者伴氨基酸尿。尿磷排泄正常或增高,尿羟脯氨酸增高。
- 血清25-(OH)D:多为正常,1,25-(OH)2D则明显升高,24,25-(OH)2D正常或稍低。
- X线骨骼检查:全身骨骼有普通性脱钙,呈典型的佝偻病骨骼X线表现,即在干骺处有增宽的透明区,干骺中心凹陷呈杯口状,其外缘向外伸展,犹如骨刺。骨皮质变薄,骨小梁粗糙,儿童患者骨化中心出现延迟。长骨骨干因矿化不全而变得边缘模糊。长骨和脊柱畸形。成人患者除有骨骼普遍脱钙和骨骼畸形外,尚有骨小梁影像模糊和假性骨折(即Loose带)或病理性骨折,但并非所有患者均有典型表现。
病因诊断依靠VDR数目/功能与基因突变检测
临床诊断HVDRR的主要依据是:①有家族史。②血钙降低,血磷正常或降低,ALP和PTH升高。③有佝偻病(或骨质软化)症状和体征,X线骨骼照片有典型佝偻病(或骨质软化)的表现。④对用以治疗VD缺乏的佝偻病的VD或其代谢产物剂量无反应,甚至一般药理剂量也无反应。
家系调查
本病为遗传性疾病,调查家庭中父母及同胞兄弟姊妹是否有同样异常对本病的诊断也有帮助,患者父母VDR基因缺陷常为杂合子。
VDR数目及其功能检测
取患者周围血分离的淋巴细胞,并用植物血凝素或病毒(杆状病毒或EB病毒)激活,然后与不同浓度的3H-1,25-(OH)2D [浓度范围为5×(10-11~10-8)M]在4℃下共同温育16小时,用吸附和离心方法,移去未结合的3H-1,25-(OH)2D,测定上清液中的放射活性,即可测出每个细胞中VDR含量(位点/每个细胞),受体对配基的亲和力为达到平衡时的解离常数。除淋巴细胞外,也可用皮肤成纤维细胞在体外培养,用完整细胞,也可用细胞质或细胞核提取物,其结果以每毫克胞浆蛋白或每微克DNA中VDR分子数表示(fmol/mg蛋白或fmol/μg DNA)。
1,25-(OH)2D诱导24,25-(OH)2D生成试验
在体外培养的成纤维细胞的媒液中加入一定量的25-(OH)D和1,25-(OH)2D,温育后测媒液中24,25-(OH)2D的含量。因为1,25-(OH)2D诱导25-(OH)D转变为24,25-(OH)2D以VDR为介导,故测定24,25-(OH)2D产量可间接测定VDR的功能。VDR有缺陷时,24,25-(OH)2D产量少于正常人相同细胞(正常人24-羟化酶活性明显增高20倍)。检测VDR结合的方法是将正常人与患者的皮肤成纤维细胞制备的可溶性提取物与3H-1,25-(OH)2D共同温育,在DNA赛璐珞上作色层析,然后用不同浓度的氯化钾溶液洗脱。正常人VDR复合物与DNA赛璐珞结合后,在氯化钾浓度为0.20~0.26mol/L时被洗脱出来;患者则在0.09~0.13mol/L时即被洗脱出来,由此证明VDR复合物与DNA结合有缺陷。同时体外试验不能检出1,25-(OH)2D是否定位到细胞核,只提示VDR的DNA结合区有突变。这种方法虽不能确定突变的确切位点与类型,但可检出在妊娠中期羊水中的来自胎儿皮肤表皮类成纤维细胞,用体外培养方法进行此项检查可对胎儿作出诊断。
通过成纤维细胞3H-1,25-(OH)2D结合试验,可鉴别出结合阴性型和结合阳性型患者。前者的成纤维细胞结合3H-1,25-(OH)2D很少,且不能诱导24-羟化酶活性。1982年以后,人们用抗VDR单克隆抗体进行此项试验,明显提高了结合试验的敏感性。Pike等发现,即使在结合阴性患者中,其成纤维细胞提取液中仍存在一种免疫反应性蛋白质,认为VDR蛋白并不减少,其缺陷不是合成障碍而是VDR的结构异常,即这种受体蛋白缺乏与1,25-(OH)2D结合的能力。
结合试验阳性型的特点是培养的成纤维细胞有结合3H-1,25-(OH)2D的能力,但无生物学反应,果糖梯度离心得到的VDR分子量正常,但在低渗盐液中不能形成聚合物,其与DNA结合的亲和力明显下降(VDR的DNA结合区突变所致)。
VDR基因缺陷检测
即利用分子生物学技术聚合酶链反应(PCR)以得到VDR受体基因的许多片段,再对每个片段测序,以检出基因的突变。将此种突变基因转染给某种细胞,其所复制的VDR与患者的VDR有相同性质,即证明此种基因突变就是被检患者的病因。为了缩小检测的分子病因范围,可根据配体结合阴性和阳性结果先分为两类。配体结合阳性,表示其突变最可能存在于VDR的DNA结合区(DBD区)。文献报道的DBD区部分突变位点见图6-37-61。相反,如果配体结合为阴性反应,表示其突变最可能存在于VDR的配体结合区(LBD区)。文献报道的LBD区突变部分位点见下图,但VDR基因的突变也可发生于交链区(如Q152stop,配体结合阴性反应),或剪接部位(如E92fs和L233fs,配体结合阴性反应)及7~9号外显子(碱基缺失)等。所以最好是对全VDR基因作突变检测。
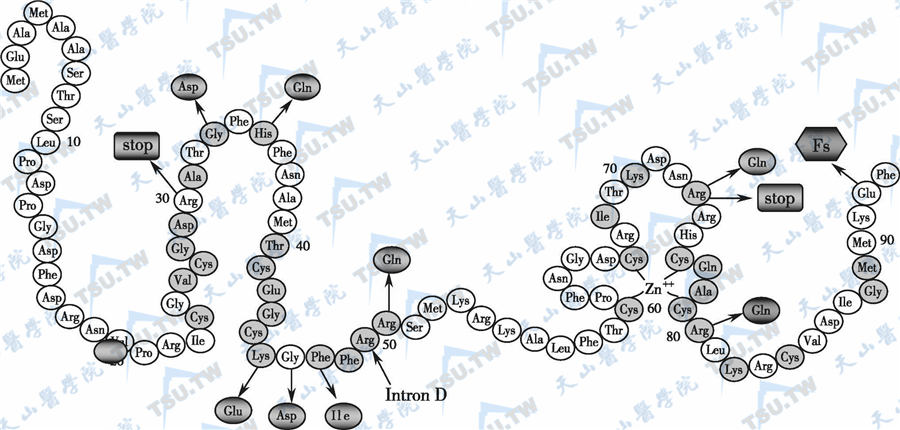
VDR的DNA结合区(DBD)及其部分突变位点的分布
注:图中显示出VDR中DBD区的两个“锌指”结构; 代表保守氨基酸残基;
代表保守氨基酸残基; 所示为突变位点;
所示为突变位点; 表示错义突变;
表示错义突变; 表示无义突变(使VDR蛋白被截短);
表示无义突变(使VDR蛋白被截短); (Fs)表示框架移动;第2、3号外显子之间为内含子D。
(Fs)表示框架移动;第2、3号外显子之间为内含子D。
HVDRR属常染色体隐性遗传性疾病,部分靶组织对1,25-(OH)2D抵抗。除典型的临床表现外,患者的皮肤成纤维细胞也存在VD抵抗,而且细胞的VDR数目减少。在24℃时,VDR与3H-1,25-(OH)2D的亲和力仅为正常VDR的1/50,VDR基因序列分析可明确其分子病因,可伴或不伴VDR的信号转导活性下降,但当用RXRα处理后,可恢复正常。
如果根据有无毛发缺失将HVDRR分为两类,那么文献报道的VDR基因突变的类型分别是:①伴有毛发缺失的HVDRR突变有:R30stop、G33D、H35Q、K45E、G46D、F47I、R50Q、R73Q、R73stop、R80Q、E92fs(读码框架移动)、Q152stop、C190W、L233fs、Q259P、Y295stop、R391C、F251C、Q317stop及7~9号外显子缺失等。②不伴有毛发缺失的HVDRR突变有:R274L、H305Q和I314S、I268T、E420K、H141、T142缺失/L141、W142和A143插入。
HVDRR与1α-羟化酶缺陷症/VD缺乏症鉴别
VD缺乏症
原发病有助于其他抗VD佝偻病/骨质软化症与HVDRR的鉴别,引起VD佝偻病的疾病很多,但均为继发性,VDR无异常,随着原发性疾病的治愈或好转,其抗VD佝偻病也随之消失或进步,血1,25-(OH)2D不升高,因而可与本病鉴别。HVDRR与VD缺乏性佝偻病/骨质软化的鉴别要点是后者可被VD治愈。在幼儿中,VD缺乏引起的佝偻病较为常见,临床上有佝偻病表现,故应与本病鉴别。VD缺乏性佝偻病有下列特点:①血钙、血磷低。②血清中24,25-(OH)2D和(或)1,25-(OH)2D降低。③无VDR缺陷。④无家族性,有营养缺乏或饮食中VD缺乏史。⑤用抗佝偻病所用的一般剂量的VD剂量治疗可以治愈。
假性佝偻病Ⅰ型
与HVDRR的鉴别依赖于血1,25-(OH)2D测定及活性VD治疗试验,假性佝偻病Ⅰ型是由于肾脏1α-羟化酶缺陷所致,临床上,但根据血清1,25-(OH)2D降低、无继发性甲状旁腺功能亢进以及对生理剂量的1,25-(OH)2D治疗有反应等可与本病鉴别。由表可见,如患儿无毛发缺失,最好的临床鉴别方法是测定血清1,25-(OH)2D及进行活性VD治疗试验。
HVDRR与1α-羟化酶缺陷症的鉴别
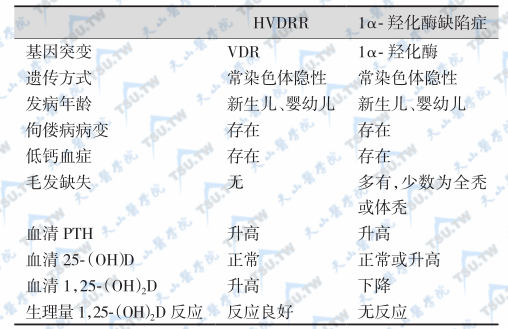
注:HVDRR:遗传性维生素D抵抗性佝偻病(或软骨病);VDR:维生素D受体
选用不同VD制剂治疗遗传性VD抵抗性佝偻病
天然VD治疗VDRR1无效,大剂量给予可能获得症状改善。但用生理剂量的1,25-(OH)2D或1α-(OH)D治疗有很好疗效,症状改善迅速。剂量个体差异大,需要根据临床症状和血清钙、磷以及ALP水平定时调整。西那卡塞特(cinacalcet)对血清PTH和骨骼代谢的作用与活性维生素D不同,可增加破骨细胞数目,以周期方式降低PTH,且不能纠正低钙血症和生长板异常。
VDRR2为遗传性疾病,缺陷在VDR基因,故尚难以治愈。但是VDR缺陷的主要后果是1,25-(OH)2D不能发挥作用而导致佝偻病的发生。VDR缺陷多为部分性,因此仍然可用1,25-(OH)2D治疗,但所用药物的剂量及反应则因人而异。
不同的VD制剂效力不同,在一般情况下,VD∶24,25-(OH)2D∶1,25-(OH)2D的效力比约为1∶10∶1000,不同的VD剂量可按此比例计算。有的作者报告每日用4000U VD2(相当于1mg),患者有良好反应;有的患者用6μg/(kg·d)的1α-(OH)D和补钙29g,血清中1,25-(OH)2D达5561.5pg/ml(正常值为20~50pg/ml),血钙仍只有1.96mmol/L,且仍有骨痛;有的患者对1,25-(OH)2D治疗反应差,而用人工合成的24,25-(OH)2D可使低钙血症得到纠正,而且在停止治疗后,血钙保持正常的时间长,这一结果颇令人费解。虽然也能与靶细胞核中的特异性DNA结合,而1,25-(OH)2D类似物的生物效应与VDR的占据量成比例,因此很难解释24,25-(OH)2D对本病的疗效反而优于1,25-(OH)2D。
因为患者1,25-(OH)2D受体缺陷严重程度不均一,故1,25-(OH)2D所用剂量不一。一般以选用1,25-(OH)2D或1α-(OH)D为好,如选用其他1,25-(OH)2D类似物或VD制剂,则需更大剂量。有的患者随着年龄增大,骨骼病变可自行缓解或愈合。如果停止治疗后,临床无症状,骨骼病变不发展,则可停止治疗。但任何年龄,只要有活动性佝偻病(或骨质软化)继续存在,治疗就应继续。VD类似物[如1,25-(OH)2D]可部分或完全纠正突变型VDR对配体的反应性,VD类似物主要用于影响配体结合的那些VDR突变(如R274L和H305E等)病例,而对干扰二聚体化过程的或辅激活子转录活性的那些突变类型的效果较差。
在用1,25-(OH)2D治疗的同时,应同时补充较大剂量钙剂,因为小肠对1,25-(OH)2D作用有抵抗(尤其是老年患者),故有钙吸收不良。一般每天需补充元素钙1000mg。
