
根据血钙/血镁/血液酸碱度鉴别手足搐搦病因
根据血钙水平,手足搐搦可分为低钙血症性和正常血钙性手足搐搦两种。
低钙血症性手足搐搦
手足搐搦的病因(酸中毒、缺氧、脑损害等)很多。首先应确立手足搐搦的类型(如局部性或全身性手足搐搦),并需与癫痫的全身性惊厥状态(generalized convulsive status epilepticus)鉴别。低钙血症性手足搐搦主要有下列3种情况:①VD缺乏引起的成人骨质软化症:血清无机磷降低或正常。X线骨片有骨质软化特征表现。②肾性骨病:肾衰竭患者虽可有低血钙和高血磷,但伴有氮质血症和酸中毒。肾小管性酸中毒患者虽血清钙降低,但血清磷正常或降低,常伴低血钾、酸中毒、尿酸化能力减退。肾性骨病虽然血清总钙降低,但因酸血症能维持离子钙浓度接近正常,很少发生自发性手足搐搦。③其他原因引起的低钙血症:饮食含钙低、消化道钙吸收不良、妊娠或骨折愈合期的钙质需要量增多,偶可伴有手足搐搦。在临床上,药物(降钙素、二膦酸盐、天门冬酰胺、普卡霉素及苯妥英钠等)引起的低血钙易于鉴别。④甲状旁腺切除术后纤维性骨炎:严重纤维囊性骨炎时,因骨矿物质缺乏而在甲状旁腺切除后血钙降低。
正常血钙性手足搐搦
引起正常血钙性手足搐搦的原因主要有呼吸性碱中毒、代谢性碱中毒、低镁血症和神经精神性疾病,根据血钙、血镁、酸碱度等容易鉴别。大多数低镁血症是由于长期营养缺乏所致;在这种情况下,低钙血症主要是由于PTH急性缺乏所致,但血磷酸盐下降(甲旁减者升高),在慢性肾衰竭中尽管有继发性甲旁亢,仍常存在低钙血症和高磷酸盐血症。
根据临床表现/PTH/25-(OH)D/血磷鉴别低钙血症病因
急性暂时性低钙血症多是急性重症疾病的一种并发症,而慢性低钙血症一般只见于几种有PTH缺乏或作用障碍性疾病。低钙血症的病因可分为甲状旁腺相关性低钙血症、维生素D相关性低钙血症和其他原因所致的低钙血症3类(下表)。对于低钙血症患者,应首先除外低白蛋白血症,并应常规测定血磷、碱性磷酸酶和尿素氮。如低钙血症伴低磷血症,血碱性磷酸酶增高而尿素氮正常,或营养不良者伴小肠吸收不良或肝脏病变时,应考虑维生素D缺乏性低钙血症可能。如血PTH增高,尿钙减少,尿cAMP增加而25-(OH)D 和1,25-(OH)2D降低,有助于维生素D缺乏症的诊断;如低钙血症伴高磷血症,碱性磷酸酶和尿素氮升高,应考虑为肾病所致的低钙血症。
低钙血症的病因鉴别

甲状旁腺相关性低钙血症
测定血钙、血磷和肌酐可做出低钙血症的初步判断。低钙血症伴高磷血症且肾功能正常是甲旁减的表现。血PTH下降,无论有无低钙血症均可诊断为甲旁减。颈部手术提示为迟发性术后甲旁减。发育缺陷,尤其是在儿童和青少年期出现的发育缺陷,提示假性甲旁减的诊断,PTH抵抗性甲旁减者血PTH增高。缺乏甲状旁腺、PTH分泌障碍或PTH抵抗所致的低钙血症一般可通过血钙、血磷、尿钙、尿磷和PTH测定得到初步诊断,因PTH缺乏和高磷血症抑制肾脏1α-羟化酶活性而使1,25-(OH)2D减低。甲旁减和假性甲旁减是终身性疾病,PTH抵抗者血PTH升高,但仍有血钙降低和血磷增高。
手术后甲旁减可发生于手术后近期,偶可于30年后首次发病。这与手术造成的局部损伤、血流障碍和甲状旁腺被毁的程度有关。无论是手术后甲旁减或特发性甲旁减,都可以在相当长的时期内呈亚临床型经过,仅在某些诱因(如月经、高热、劳累、寒冷和情绪改变等)下诱发手足搐搦。
遗传性甲旁减的发病较缓慢,继发性甲旁减没有发育缺陷。两者均可有基底核钙化和锥体束外综合征,这在遗传性甲旁减中更为常见而且出现较早。两者均可有视神经乳头水肿和颅内压升高,指甲、毛发的慢性改变以及晶状体白内障。在遗传性甲旁减中,还有某些特殊的皮肤表现(如脱发和念珠菌病)。假性甲旁减是由于PTH作用障碍所致,但该病也具有甲旁减的某些临床特点,包括骨外钙化和锥体束外综合征,如手足搐动、肌张力障碍等。在某些情况下,继发性甲旁减并不是因甲状旁腺组织被切除,而是由于手术后颈部发生的纤维化影响了甲状旁腺的血液供应或其他一些仍未明了的原因所致。
维生素D相关性低钙血症
维生素D缺乏症、维生素D抵抗综合征和1,25-(OH)2D生成障碍或维生素D丢失过多引起低钙血症。低钙血症伴正常或降低的血磷时,应测定VD。成人新近发生的低钙血症一般是由于营养缺乏、肾衰竭或肠道疾病所致。VD水平正常或升高而伴有佝偻病/骨质软化和各种神经肌肉综合征以及骨畸形提示VD抵抗性甲旁减。
其他原因所致的低钙血症
主要包括:①钙盐沉积于骨过多(成骨细胞性肿瘤、骨饥饿综合征);②钙螯合剂(Foscarnet、磷酸盐、EDTA、氟制剂)和抗惊厥药物;③新生儿低钙血症(早产儿,母亲患甲旁减、甲旁亢或糖尿病);④HIV感染(抗HIV药物、维生素D缺乏、低镁血症、PTH抵抗);⑤急性疾病(急性胰腺炎、中毒性休克等)。
根据临床资料鉴别甲旁减的病因与类型
综合分析临床资料是鉴别甲旁减的病因与类型的基本方法,这些资料主要包括血钙、血磷、血镁、PTH、25-(OH)D、尿钙、尿磷和影像检查等,偶尔还需要结合动态试验与遗传学及基因突变分析才能做出鉴别。
自身免疫性多内分泌腺病综合征
其特点是同时或先后发生两种或两种以上的内分泌疾病。除了甲状腺可有功能亢进(Graves病亦属自身免疫病)外,其余多属功能减退。在157例Ⅰ型自身免疫性多内分泌病综合征患者中,白色念珠菌病者占73%,甲旁减占88%,慢性肾上腺皮质功能减退者占59%,秃发占20%,性腺功能早衰占40%。在患者及部分家属中,可检出血清甲状旁腺抗体。受损内分泌腺的病理特点是淋巴细胞浸润及纤维化。有些报道的病例合并恶性贫血及腺垂体功能减退症。
甲旁减合并肾上腺皮质功能减退症常发生于儿童,尤多见于1~6岁。甲旁减往往比Addison病发生得早。88%的患者在10岁前出现甲旁减,其后2~4年内Addison病随之发生,女性略多于男性。若先证者患甲旁减合并Addison病,则其兄弟姊妹发生甲旁减或Addison病的概率为35%。若累及甲状旁腺,则甲旁减的临床表现、诊断方法与治疗与特发性甲旁减相同。有条件者可查血液抗甲状旁腺抗体。甲旁减合并Addison病或其他内分泌腺疾病时,其还应包括Addison病或其他内分泌腺疾病的诊断。
假性特发性甲旁减
假性特发性甲旁减综合征是指分泌的PTH生物活性降低,临床表现亦是低钙血症,与特发性甲旁减之临床表现相同。其实验室检查结果与特发性甲旁减相同,但用放射免疫法测量时PTH正常或升高。鉴别的重点是前者有特殊体型、甲状旁腺形态及功能正常或增高。在X线平片上假性甲旁减表现为骨骺早期愈合。掌(跖)骨及指(趾)骨发育短,严重者呈矩形,常以第1、4、5掌骨和第1、4跖骨最明显,两侧可对称或不对称(图3-14-23)。指(趾)骨也变短,以中节指骨增粗为主,末节指骨短于正常,可呈三角形。掌骨征阳性,表现为手部正位摄片时,在第4与第5掌骨头远侧顶端划一连线并向桡侧延伸,该延长线与第3掌骨相交(足部可有类似表现)。正常人该线超越第3掌骨头而不与其相交。该征最多见于该病患者,但亦可见于少数正常人及长骨粗短、骨皮质增厚、短指、桡骨弯曲、髋内(外)翻畸形、外生骨疣及Turner综合征患者,结合其他征象不难与该病鉴别。
假性家族性甲旁减
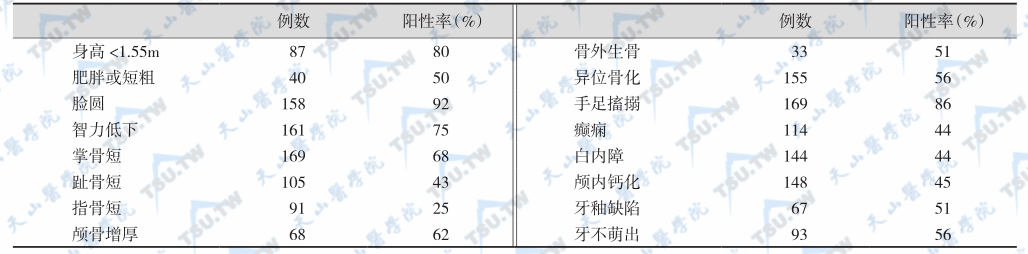
是一种罕见的家族性甲状旁腺疾病,X伴性显性、常染色体显性或隐性遗传,伴多种类型的先天畸形及缺陷(包括躯体、感觉器官及内分泌腺缺陷)。周围靶器官受体或受体后缺陷,对PTH无反应。临床表现为甲旁减,患者具有甲旁减的低血钙、高血磷、手足搐搦及尿钙磷变化等特点,但甲状旁腺增生,PTH分泌增多。可有智力减退并呈特殊的体态,如身材粗矮、肥胖、圆脸、颈粗短、指(趾)短小畸形,常见第1、4、5掌骨或跖骨缩短,以致握拳时在1、4、5掌骨头部形成凹陷(Albright征)。假性甲旁减临床上还常有味觉、嗅觉障碍等,可合并甲状腺功能减退、肾上腺皮质功能减退、尿崩症、糖尿病、性腺发育障碍或不发育。下表列举了假性甲旁减的症状及体征发生率。
假性甲旁减的症状及体征
家族性Fahr综合征
亦称为对称性大脑钙化综合征(symmetrical cerebral calicification syndrome),常伴血卟啉病、顽固性贫血、假性甲旁减(2型),血清转铁蛋白显著升高、血清铁和铁结合力下降和铁沉着症,双侧对称性基底核钙化,小脑齿状核和脑沟处亦可见钙化、基底核钙化症、大脑钙质沉着症、家族性基底核钙化症、家族性特发性基底核钙化症、特发性家族脑血管亚铁钙沉着症、特发性家族性脑血管铁钙质沉着症、特发性非动脉硬化性脑血管钙化症、大脑钙质沉着伴晚发性脑病、特发性两侧对称性大脑基底核钙化症等。
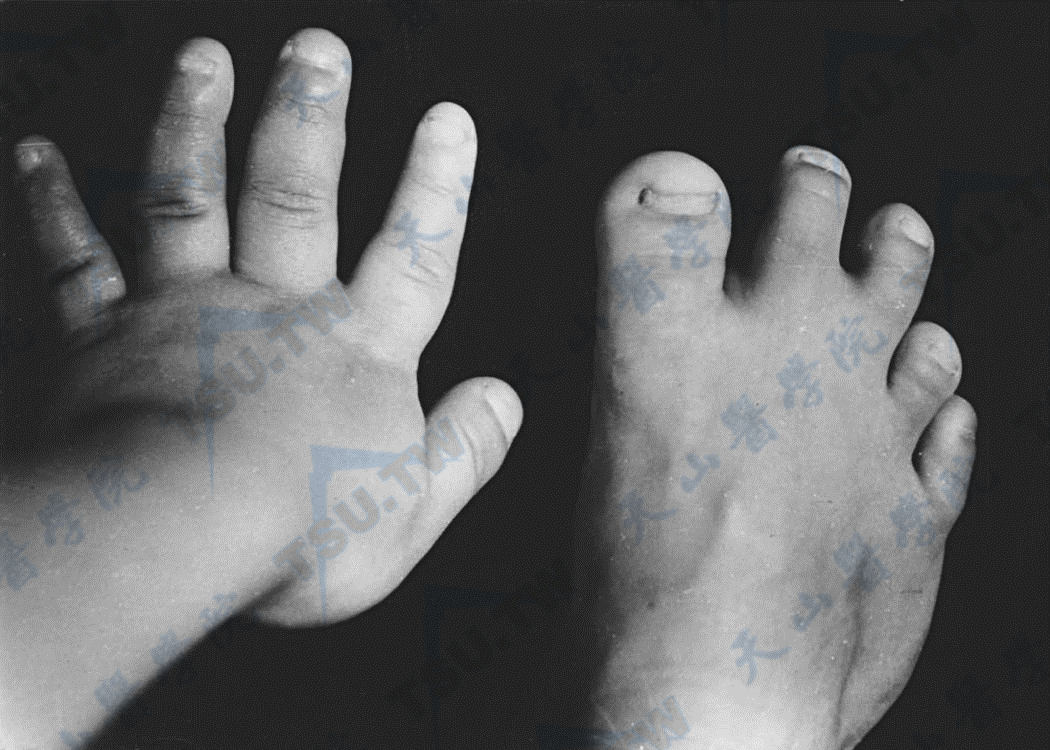
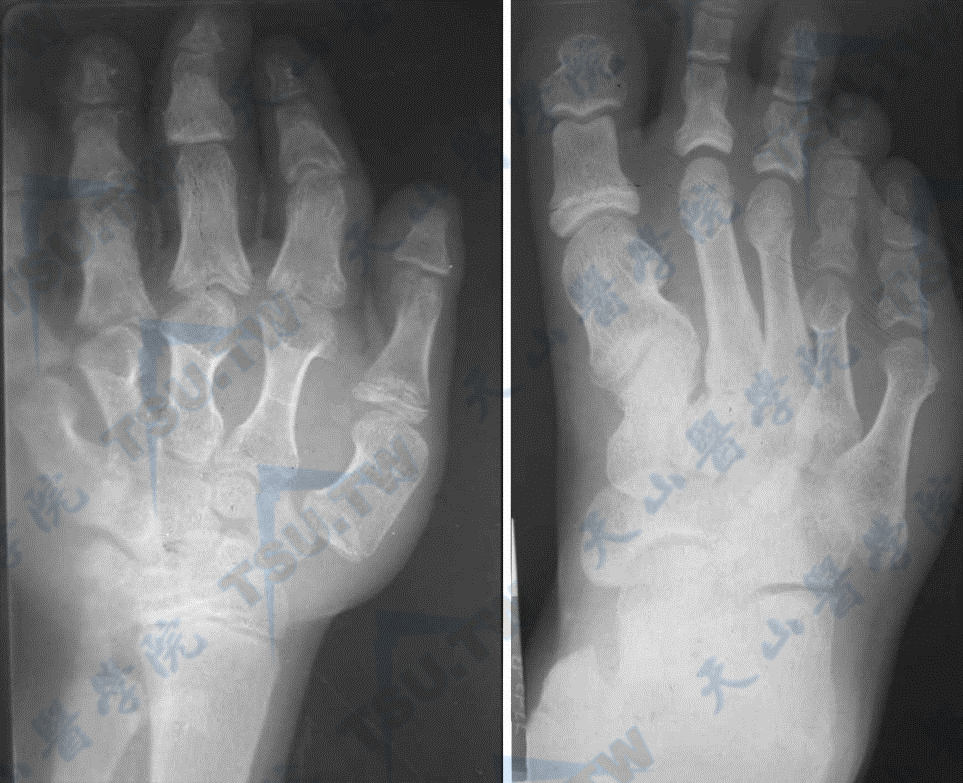
注:女,14岁,假性甲旁减Ⅰ型。左手、右足像片及X线平片示左手掌骨及手指骨粗短,第1掌骨,第2、3、4近节指骨有锥形骨骺;右足跖、趾骨亦短,第1、4、5跖骨及趾骨粗短较显著
本征常在脑CT等检查时发现双侧对称性基底核钙化,小脑齿状核和脑沟处亦可见钙化。一般不伴有症状。随着年龄增长到老年前期(45~60岁)时,可表现不同程度的神经症状,如精神衰退、癫痫发作、小脑性共济失调、情绪迟钝、记忆减退、类精神病样症状、帕金森病、构音障碍等。Billard等总结具有脑病表现的14例患者的临床表现,认为有4种临床类型:
- 第1型呈常染色体隐性遗传,病理改变以神经脱髓鞘和钙化为特征,表现为脑病、小脑畸形(microcephaly)、矮小、视网膜病变和视交叉萎缩。本型发病较早,进展较快;
- 第2型有先天性脑病或脑瘫表现,但无短肢畸形,亦无眼部和脑脊液异常;此型很可能不是遗传性疾病,估计与产前的病毒感染有关;
- 第3型有脑病和小脑畸形,脑脊液中淋巴细胞持续增多;
- 第4型的主要表现为基底神经节钙化,伴或不伴神经功能异常,本型呈常染色体显性遗传。
甲旁减-发育延迟-畸形综合征
甲旁减-发育延迟-畸形综合征(hypoparathyroidism-retardation-dysmorphism syndrome,HRD)多见于儿童,表现为甲旁减、感觉神经性耳聋、肾发育不良、基底核脑梗死等。HRD还可出现反复发生的基底核脑梗死。染色体检查显示有del(10)(p14-15.1)异常,提示与HDR综合征有关的基因定位于10p14-15.1。2000年,van Esch等发现此染色体上GATA3突变是HRD发病的分子基础 。
Di George综合征
Di George综合征又称腮发育异常症(branchial dysmorphogenesis),第Ⅲ-Ⅳ咽囊综合征(thirdfourth pharyngeal pouch syndrome)或先天性胸腺发育不良,是人类最常见的微缺失综合征,Di George综合征是其中一种重要的类型。有5%~10%的患者具有Di George综合征的临床表现,但不存在22q11.2缺失。Di George综合征可出现80余种先天性缺陷。常见表现为无甲状旁腺及胸腺,有心脏异常、面容异常及其他畸形。由于缺乏胸腺,患者免疫缺陷,抵抗力很低,常有感染。尚可合并第1对腮弓发育缺陷造成的畸形,表现为两眼距离宽、眼角上斜、两耳低或不对称、短人中及小下颌。若合并第5对腮弓发育不良,则可发生主动脉右位或法洛四联症。出生后即有低钙血症,手足搐搦。可有先天性心脏病和其他畸形,因易感染,常易发热,甚至夭折。患者有低钙血症和高磷血症,碱性磷酸酶正常,PTH减低,免疫功能低下。此外,患者还可出现行为异常和学习困难等。
Kearns-Sayre综合征
极少见。国外于1958年首次报道,我国于1988年首次报道。该病以进行性眼外肌麻痹、色素性视网膜病和心脏传导阻滞为主要特征,还可出现中枢神经异常和特发性甲旁减等表现。神经系统病变进展缓慢,心脏异常的主要表现以传导系统受累且是早期死亡的主要原因。Kearns-Sayre综合征属线粒体脑肌病,线粒体DNA发生缺失或点突变,不能编码线粒体在氧化过程中所必需的酶或载体,糖原和脂肪酸等原料不能进入线粒体,或不能被充分利用,故不能产生足够的ATP而导致能量代谢障碍。如肌肉活检见破碎红纤维,电镜下线粒体异常,线粒体呼吸链酶异常,DNA分析发现mtDNA缺失或点突变则可确诊。早期易误诊为重症肌无力、眼肌型或眼咽型进行性肌营养不良症、眶后肿瘤或脑垂体瘤、周期性瘫痪。慢性进行性眼外肌瘫痪、眼底检查及心脏损害的出现有利于鉴别。目前无特效治疗,给予ATP、辅酶Q10、大量B族维生素,及早植入起搏器可延长生命,低钙血症的治疗与特发性甲旁减相同。基因治疗是今后的发展方向。
骨饥饿综合征
骨饥饿综合征(hungry bone syndrome)是指骨钙丢失十分严重的患者在手术等治疗之后,由于血钙急剧向早已有钙严重缺乏(饥饿)状态的骨组织转入,以致血钙下降引起手足搐搦等。不仅血清钙、血清磷下降,尿中钙、磷也减少,尿中羟脯氨酸术后一过性减少,而后再度增多,血清ALP升高。典型病例见于骨型的原发性甲旁亢的甲状旁腺摘除术后,或甲状腺功能亢进症的甲状腺次全切除术后。通常情况下,术后低钙以第2~3天最为严重,但如果症状不明显,第4~5天后会自然缓解,如果术后血钙低于1.9mmol/L且有搐搦发生,则应在术后第1~2天静脉补钙,对于持续时间长、补钙效果差的术后患者应考虑骨饥饿综合征的存在。
磷酸酶缺陷综合征
本征为一种异质性疾病,由于组织特异性碱性磷酸酶(TNSALP)活性不足所致。TNSALP的编码基因位于1p34-36.1,该征有两种遗传模式。先天型为常染色体隐性遗传,为致死性疾病。常染色体显性遗传者病情较轻,发病较迟。依据年龄、临床表现及X线所见,临床分3型:①新生儿型:可在出生前发生骨折、长骨弯曲畸形、串珠肋、囟门扩大、骨端肥大等,多在1年内夭折。②儿童型:常以生长发育落后就医,X线表现似佝偻病。③成人型:青年或成年发病,表现最轻,少见。常因轻微外伤或关节病引起骨折而被发现,或常有肾结石、出牙困难及生长发育迟缓历史。生化特点是血清钙偏高,血磷正常,血清碱性磷酸酶低下。尿钙增多,尿中排出大量的磷酸氨己醇,羟脯氨酸减少。X线典型表现为颅骨和椎骨骨化减弱或颅盖骨显著变薄。其他骨改变包括管状骨变短并且骨化不良或骨化不规则,干骺端类似佝偻病样改变。诊断时尚需与维生素E缺乏、甲减、镁缺乏等鉴别。本征目前无特效疗法。
IgM缺乏症
常与22q11微缺失综合征并存,除有22q11微缺失综合征的一般表现外,患者常以反复发作的慢性中耳炎或发育延迟而就诊。
