
低磷酸酶症是一种罕见的代谢性骨病。于1948年由John Campbell Rathbun首先报道。50多年后,Mumm等从Rathbun报道的婴儿病例的父母的DNA分析中发现,当年他报道的病例的分子病因为TNAP基因两个错义突变(G340A/A881C)。至此,报告的病例达300余例,世界范围内均有发病,所有种族均可受累,通常为常染色体隐性遗传,少数患者有父母近亲婚配史。临床上以骨矿化不足、血清ALP降低以及尿和血液中磷酸氨基乙醇(phosphoethanolamine,PEA)含量和血钙升高为特征。多数患者在婴儿或儿童期即可出现症状,如颅骨未骨化或软化,囟门增大并闭合迟延;有些患者因症状轻微可迟至成人期才被发现。严重者通常在出生后不久即夭折。轻型的儿童病例可仅表现为学步延迟和乳齿早脱。
成人患者多因轻微外伤性骨折首次就诊,骨折难以愈合。X线表现为膜化骨和软骨内矿化不足或缺乏。颅骨无矿化,管状骨骨折并畸形,身材矮小和四肢短小畸形。存活的婴幼儿可有不同程度的骨骼受累,主要在骺板和干骺区,其改变与佝偻病相似。成年患者可表现为骨质软化,有时还可见假性骨折(looser带)和少量骨膜下新骨形成。
TNAP功能异常导致低磷酸酶症
本病是由于TNAP基因突变所致。至目前为止,已发现突变基因类型八十多种。突变类型多为错义突变,少数为复合性突变和无义突变。在欧美,以TNAP基因的E174K突变为最常见;在日本,等位基因的缺失频率高达36%。
除假性低磷酸酶症ALP活性正常外,其他类型的低磷酸酶症ALP活性均降低,严重者无法测出。虽然TNAP分布于全身所有的组织中,但TNAP基因突变只引起骨和牙组织病变。Iqbal等分别测定低磷酸酶症的TNAP的免疫活性和生物(催化)活性,得到两者的回归关系式为cBALP(骨源性ALP催化活性)=0.796+3.269iBALP(骨源性ALP免疫活性)。一般iBALP和cBALP越低,骨骼病变也越严重。如果血清的总TNAP正常,还可测定中性粒细胞ALP(neutrophil ALP,NAP),得到的计数值(正常人为20~150)如低于15则多为杂合子突变患者。同样,测定ALP同工酶也有相当价值,凡有骨骼病变者,骨源性ALP将显著降低。另一方面,肝型ALP降低者,可无临床表现。
- 第一类突变:如R54C、R54P、A94T、R206W、G317D 和V365I使TNAP活性完全丧失,于宫内或出生后不久夭折。
- 第二类突变:如A16V、A115V、A160T、A162T、E174K、E174G、D277A、E281K、D361V和G439R使TNAP的活性部分丧失,但严重程度可有较大差别,有些突变仅使酶对一个底物的催化活性丧失,可仅有牙组织的TNAP活性下降,而对另一底物的催化活性完全正常。如A160T突变时,催化对-硝苯磷酸盐(p-nitrophenylphosphate,PNPP)的活性下降,而对PPi的催化活性正常。而D227A突变后的活性变化与A160T刚好相反。
- 第三类突变:如E174G、E174K、E281K使酶对PNPP 和PPi的催化活性均正常,但对PLP无催化活性,使PLP在脑组织中堆积中毒,导致癫痫样发作。
TNAP突变主要发生于分子链的5个关键区段,即活性位点(active site)及其邻近活性位点凹陷区(active site valley)、同二聚体界面(bomodimer interface)、冠状结构域(crown domain)和金属离子(Ca2+)结合位点(mental-binding site),其中后两种结构域为哺乳动物胎盘ALP(placental ALP)所特有。冠状结构域含有胶原结合环(collagen binding loop)。一般引起酶活性显著下降的突变多位于Ca2+结合位点及其附近。临床症状取决于突变部位及程度。
成骨细胞分泌的ALP和浆细胞膜型糖蛋白-1是两种相互制约的调节因子,TNAP活性不足,堆积的PPi抑制羟磷灰石结晶的形成和生长。浆细胞膜型糖蛋白-1(plasma cell membrane glycoprotein-1,PC-1)仅存在于膜限制性基质囊泡(membrane-limited matrix vesicles,MVs)的内侧面,PC-1基因突变小鼠表现为矿化过度,导致骨关节炎和脊椎后纵韧带钙化,而PC-1基因和TNAP基因均缺失的小鼠又表现为正常的矿化功能。因此,凡能抑制PC-1功能的举措都有可能成为治疗低磷酸酶症的途径。
低磷酸酶症分为六型
- 围产型(先天型):均为死产或产后不久死亡,表现为骨矿化不良,四肢短,先天性矮小,有的死产儿几乎无骨的矿化。
- 婴幼儿型:有生长障碍,常因抵抗力低而并发肺炎,并常出现高钙血症和高钙尿症,身材矮小,呈佝偻病样畸形;严重病例常伴频发性维生素B6依赖性惊厥和癫痫样发作,维生素B6补充可终止发作。
- 儿童型:出牙迟,牙齿发育不良,儿童型低磷酸酶症(childhood-type hypophosphatasia)的表型多变,乳牙过早脱落为其常见和特征性表现。肢体短(可低于正常的2s),肌张力低下;前额增大,全身骨的矿化延迟落后,伴肾石病或肾钙盐沉着症,BMD降低。全身生长发育不良,体重低,有些患者可伴有颅缝早闭(craniosynostosis)。TNAP活性可降至正常的5%以下。学步晚,青春期发育延迟。
- 成年型:软骨发育不良,反复发生应激性骨折,造成肢体畸形,中年以后发病者少有佝偻病病史。
- 牙型:仅有牙齿的表现。由于牙冠矿化受阻,使牙槽骨间隙小,牙齿生长异常。Watanabe等报道,TNAP的杂合子突变(第9号外显子的T1155C突变来自母方而第10号外显子的G1320A突变来自父方)表现为牙型低磷酸酶症,伴严重牙周炎和牙病变。牙槽和牙发育不良,除乳牙早脱外,还可伴有颌骨异常。在光镜、偏振光显微镜和扫描电镜下,可见牙根有大量的骨吸收灶,尤以底部突出,牙本质矿化不良,牙骨质(root cementum)发育不良或无发育,根部牙本质亦有吸收。
- 假性低磷酸酶症:与上述儿童型、成人型的临床表现及X线表现相同,但血ALP正常。
一些观察结果提示,上述的分类方法可能过于粗糙,各型中还包括不少的亚型。例如在围产型中,Pauli和Moore等报道5例患者有严重骨发育不良,酷似重型成骨不全。但出生后的病程呈良性过程,有长骨发育,其他病变亦有自发缓解趋势,称为良性胎儿型低磷酸酶症(benign prenatal form of hypophosphatasia)。
ALP底物堆积导致特异性表现
骨畸形、骨折和抵抗力低下是导致预后不良的主要原因。婴幼儿患者常并发肺炎、高钙血症、骨畸形和癫痫样发作。儿童患者易并发肾石病、肾钙盐沉着症和颅缝早闭。
- 头颅:严重者头颅增大、前额突出,所有颅骨均无矿化。晚发病者的颅缝早闭,因颅内压增高引起颅骨内板指样压迹增多、加深,小头畸形或见缝间骨以及乳齿过早脱落。
- 脊柱:严重病例的矿化椎体可薄如纸片,椎弓亦无矿化。椎旁可见斑条状矿化或钙化影。
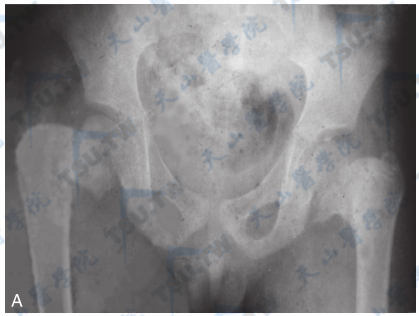
- 胸廓及骨盆:胸廓及骨盆狭小或伴软化畸形,矿化不良,甚至或完全无骨化;有时可见骨盆骨有假性骨折(下图)。
- 管状骨:婴幼儿患者的管状骨粗短、弯曲畸形,伴或不伴陈旧性骨折,骨量减少,BMD减低,并可见Looser带;如有外伤性骨折,骨痂生长较少,愈合缓慢,干骺端有大小不等的不规则形骨质缺损(未钙化的骨基质),随年龄增长,干骺端则留有不规则性骨硬化改变及稀疏粗糙的骨小梁。
- 骨成熟:骨骺骨化中心延迟出现,而骨骺线的闭合可提前。ALP活性不足,ALP底物在血浆中和组织中堆积,其中焦磷酸盐(PPi)过多促进了关节软骨钙盐沉着症(articular chondrocalcinosis),这种病变主要发生于成人型低磷酸酶症患者(常染色体隐性遗传)中。
低磷酸酶症
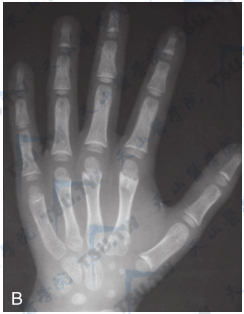
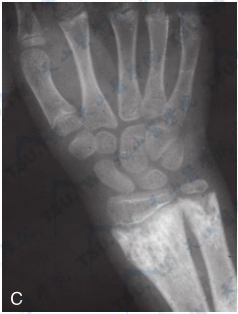
注:A:男,6岁,低磷酸酶症的骨盆及手正位片。骨盆广泛骨密度降低,干骺端变形,颈干角变小;B:手部诸骨轻度骨密度降低,第1掌骨近侧,第2、3、4掌骨远侧及远节指骨近侧干骺端呈杯口状凹陷、有粗糙骨小梁和不规则密度减低区。C:女,11岁,低磷酸酶症右腕正位片。示诸骨有轻度骨密度降低,第2、3、4掌骨、尺、桡骨远侧,第1掌骨近侧干骺端呈杯口状凹陷,边缘外展,有粗糙骨小梁和不规则密度减低区,以尺骨较重。
根据临床表现与X线检查确立诊断
低磷酸酶症的特征性表现主要为:
- 婴幼儿、儿童有生长发育迟缓,学步晚和出牙迟。
- 身材矮小、肢体短小畸形、易发生骨折或具有佝偻病样体征(如囟门未闭或闭合延迟、肋骨串珠、弓形腿等),管状骨粗短、干骺端处有大小不等的不规则骨质缺损等表现。
- 仅有牙齿发育不良,而血清ALP活性降低,ALP和PEA增高者(提示为牙型低磷酸酶症)。
- 有上述临床表现,血ALP和血、尿PEA增高,且能排除佝偻病和骨质软化症者(提示为假性低磷酸酶症)。
本症的临床确诊依据是:成人患者如无明显佝偻病后遗症状者,而易反复发生应力性骨折,且骨折难以愈合,X线表现有骨质软化(如骨小梁和骨皮质模糊不清)和骨畸形,血清ALP降低,PTH正常而血钙高、血磷低、PEA高亦可确诊。病因诊断有赖于TNAP基因的DNA分析和TNAP基因突变的鉴定。Mumm等用变性梯度凝胶电泳法(denaturing gradient gel electrophoresis,DGGE)对TNAP基因进行综合性突变分析(comprehensive mutational analysis),使用的引物和PCR扩增条件包括TNAP基因中的2~12个外显子和邻近的剪接位点,扩增子(amplicon)可掺入GC夹(GC clamp)的一端,操作简便而迅速。本法探查点突变的有效率达100%,缺点是可能遗漏大片段缺失性突变。
儿童型常有高钙尿症而无高钙血症。尿磷酸氨基乙醇受年龄和饮食的影响。磷酸氨基乙醇(phosphoethanolamine,PEA)正常值范围:<15岁为83~222μmol/g肌酐;15~30岁为42~146μmol/g肌酐;31~41岁为38~155μmol/g肌酐;>45岁为48~93μmol/g肌酐。血清TNS-ALP辅酶PLP(吡哆醛-5'-磷酸)常增高。此为较特异而敏感的指标(包括假性低磷酸酶症)。但测定前一周应停服维生素B6。血1,25-(OH)2D和PTH一般正常,个别患者可见血PTH增高,可能与肾功能不全有关。全身骨骼BMD或脊椎BMD正常或稍低,桡骨和股骨BMD可增加,由于软骨的矿化障碍,可出现骨松质的代偿性矿化过度或骨质硬化,用连锁DNA标记法进行TNAP分析,可发现突变基因。
从15周起,可行脐穿刺(cordocentesis)采样或羊水检查,测定ALP活力和ALP基因突变检查,如获得确诊,应终止妊娠。相反,如果虽然父母双方为突变的TNAP基因携带者,但绒毛膜采样未查出异常,且胎儿的骨骼发育与超声检查正常,可判断为健康者,从而避免了错误决策。
骨的非肿瘤性浸润性疾病和发育不良性疾病很多,当从X线照片中发现存在骨的这些病变后,一般要用MRI进一步了解骨病变的微细特征,尤其要特别注意局部骨髓的浸润情况,有助于早期发现此类代谢性骨病。
超声检查主要用于父母为本病携带者的胎儿产前检查。围产型低磷酸酶症(perinatal hypophosphatasia)为本病的致命型,呈常染色体隐性遗传,主要累及骨组织,使骨的矿化障碍。发病率为1/10万,为TNAP同工酶基因突变所致。本病可被早期诊断,约于妊娠12~14周时,超声检查可见项线透光带增厚(nuchal translucency thickness),颅骨和脊椎骨矿化不良,胸腔狭窄和肢体短小等改变。
低磷酸酶症与颅锁发育不良/成骨不全/软骨不发育/营养不良性佝偻病鉴别
低磷酸酶症应与颅锁发育不良症(cleidocranial dysplasia,CCD)鉴别。CCD是一种常染色体显性遗传性骨发育异常综合征,其临床特点是锁骨发育不良,前额突出,肢体缩短,牙异常和骨的其他畸形,是由于RunX2(Cbfa1)基因突变(如A169P),而TNAP基因并无变异。但一些CCD患者的血清ALP活力下降,尿中磷酸氨基乙醇(PEA)升高,并可伴有BMD降低。患者经治疗后这些异常仍可持续存在,其表现酷似低磷酸酶症。CCD患者出现低磷酸酶症表现(可称为继发性低磷酸酶症)的原因尚未明了,可能是RunX2基因突变致骨的早期成熟和骨的转换异常或TNAP基因表达下调所致。
成骨不全(婴儿型和先天型)可有颅骨矿化不良、身材矮小、长骨弯曲、短肢等,与低磷酸酶症的围产型和婴儿型相似,但成骨不全具有蓝色巩膜、骨质疏松及血ALP增高。超声检查可发现胎儿的先天性低磷酸酶症,在超声图上低磷酸酶症表现为胎儿骨骼呈普遍性钙化不良、肢体短小,围产型低磷酸酶症必须与成骨不全Ⅱ型(ⅡA和ⅡC)及软骨不发育症ⅠA鉴别,肢体骨出现“骨刺”支持低磷酸酶症的诊断。如高度疑为本病,可行脐带穿刺(cordocentesis)取血测定ALP(本病患者明显降低或不可测出)。
营养不良性佝偻病轻者表现为肢体弯曲畸形,X线有矿化不良等表现,与儿童或婴幼儿型低磷酸酶症易混淆,但佝偻病者血钙偏低、尿钙低、血ALP增加(或正常),而低磷酸酶症患儿血ALP活性减低、血钙高、血磷低及尿钙明显增多。
本病呈部分自限性,有些患者可自愈。病情较重者应采用综合性治疗。曾有学者采用静脉滴注几种具活性的碱性磷酸酶治疗,虽有生物化学上的改善(如血钙、磷、ALP基本正常),但随着酶在体内降解,活性消退,半年观察未见有放射学的改变。降钙素和氯噻嗪可纠正高钙血症,抑制骨钙释放和细胞外液钙转移。常用的有益盖宁或密钙息,用量根据病情及血钙水平确定。氯噻嗪能减轻高钙尿和骨的低矿化,从而间接降低高血钙。常用量为每次25mg,25~75mg/d。非甾体类固醇性消炎止痛药有助于缓解患者的疼痛(抑制前列腺素E的合成和释放)。因制动或肌无力,患者少活动而使机体抵抗力降低,特别是婴幼儿或严重病例有50%以上易并发肺部感染而死亡,故应补充足够的蛋白质、糖类及微量元素和维生素,以满足生长发育的需要,个别行走不便的患儿应注意被动按摩和关节的屈曲运动,同时加强看护,以免碰撞跌倒造成骨折。在动物实验中,维生素B6可治疗TNAP基因敲除(-/-)小鼠的癫痫样发作。同样,正常小鼠如严重缺失维生素B6,又可诱发如同TNAP基因敲除鼠相似的癫痫发作,但骨的代谢与成骨细胞功能不受影响。故可认为骨骼组织外的许多病变与维生素B6代谢失常有关,而骨病变与维生素B6代谢异常无关。维生素B6可诱导本病患者(如TNAP基因1154-1156杂合缺失)的紫外线过敏,其原因可能与维生素B6代谢异常有关。
