
Paget骨病(Paget disease of bone,PDB)是老年白种人中仅次于骨质疏松的常见代谢性骨病。PDB又称变形性骨炎或畸形性骨炎(osteitis deformans),是局部骨组织的一种骨重建(bone remodeling)亢进性疾病。其病变特点是病灶处的骨重建(骨吸收、骨形成和骨矿化)增加;由于过高的破骨细胞活性及过多的破骨细胞引起高速骨溶解,并导致成骨细胞增加和骨形成过多,形成的交织骨结构脆弱;骨盐及胶原的转换率增加,骨髓纤维化和血管过多致骨局限性膨大。由于骨形成和骨吸收之间失衡导致骨面积增大和骨畸形。Paget骨病可能包括了数种不同的临床类型。
Paget骨病是仅次于骨质疏松的常见代谢性骨病。发病无性别差异,初次就诊年龄多在40岁以上(尚未见有18岁以下的病例报道)。本病具有家族遗传特点,有阳性家族史者一般约占15%,最高达40%。Paget骨病主要流行于盎格鲁撒克逊人(Anglo-Saxon)后代中。据考证,音乐圣师——路德维希·凡·贝多芬(Ludwig van Beethoven,1770~1827年)患的也是Paget骨病。由于过高的破骨细胞活性及过多的破骨细胞引起高速骨溶解,导致成骨细胞增加和骨形成过多,骨髓纤维化和血管增生导致局限性骨膨大和骨畸形,并引起贝多芬失聪。
本病的流行具有显著的地理和人种特异性。例如,当单骨性Paget骨病的印度男性患者迁移到Paget骨病流行区后,其病情明显加重。Paget骨病的高危环境可能主要是病毒感染或其相关因素。本病的地域分布明显,英国的英格兰和威尔士地区最常见,发病率在55岁以上人群中约为0.3%,随着年龄增加,发病率急剧上升。考古分析公元前1850年~公元前900年古英格兰北部居民(2770例)的Paget骨病患病率,经X线照片证实者为15例,患病率为2.1%(40岁以上者)。公元前1500年间为1.7%,而公元后1500年间为3.1%,说明本病在过去的几千年中患病率增加。澳大利亚、新西兰、南非及美国次之,美国Paget骨病的总患病率不低于1%,有的地区超过2%。但近几年有证据表明,英美两国的发病率正在下降,下降的原因可能与环境因素有关。在北欧、中东阿拉伯、中国、日本该病少见。在法国、意大利及西班牙等国,发病率居中。我国和亚洲地区少见,北京、河北、河南、山东、湖南、甘肃及台湾等省均有报道。
病因和发病机制未明,但近年来有重大的新发现。从目前的研究结果看,Paget骨病很可能是一种以局限性高速骨溶解为特征的临床综合征,而高速骨溶解的基本原因是OPGRANK-RANKL信号分子或相关基因的突变。病毒感染、内分泌功能紊乱和自主神经功能紊乱也在Paget 骨病的发病中起了重要作用。因此,Paget骨病是一种基因与环境相互作用而导致疾病的典型例子。
OPG及相关基因突变引起局限性高速骨溶解
本病的家族聚集现象明显,家族性Paget骨病以常染色体显性方式遗传。近年的研究发现,Paget骨病很可能是一种遗传综合征。全基因组扫描发现了4个易感基因或遗传位点。Paget骨病样家族性扩张性溶骨症(PDB-like syndromes of familial expansile osteolysis)、早发性家族性Paget骨病(early-onset familial PDB)和扩张性骨性高磷酸酶血症(expansile skeletal hyperphosphatasia)的分子病因分别为破骨细胞的调节因子RANK插入突变(TNFRSF11A)、护骨素(osteoprotegerin,OPG)失活性突变(TNFRSF11B)和 RANK配体(RANKL)多态性有关。
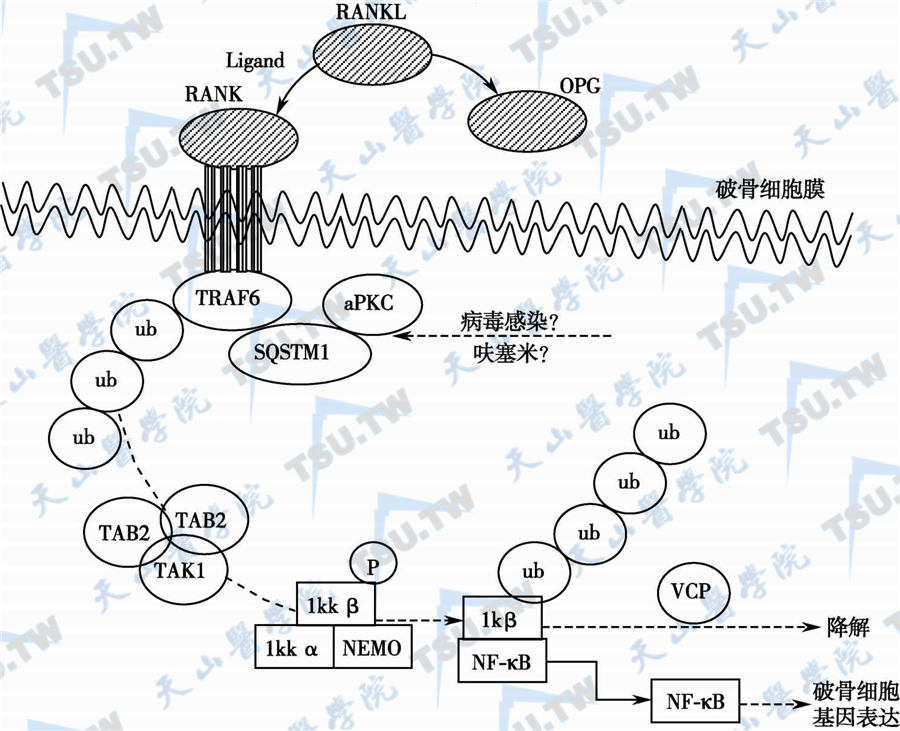
RANKL-NF-κB信号途径
注:AANKL激活RANK,引起受体蛋白三聚化,受体的胞质尾部与适应蛋白TRAF6结合,并与非典型蛋白激酶C及SQSTM1形成复合物;TRAF6在泛肽连接酶的作用下,与多个小分子泛肽结合并形成多泛肽链。下游TAB2-TAB3适应蛋白识别并结合泛肽链,并进一步将IKK2-IKKβ-NEMO复合物激活(磷酸化),后者进入核内,选择性刺激破骨细胞基因表达。OPG可阻滞RANKL与RANK结合,而IκB的降解与VCP有关。病毒和其他环境因素如呋塞米诱发Page骨病的机制未明,可能与损害SQSTM1的正常功能有关。aPKC:atypical protein kinase,非典型性蛋白激酶;SQSTM1:sequestosome1。
Paget骨病致病基因可能还包括SQSTM1以及SQSTM1相关基因。此外,含valosin蛋白(valosin-containing protein,VCP)基因突变除引起Paget骨病外,还导致遗传性包涵体肌病综合征(syndrome of hereditary inclusion body myopathy)和前颞型痴呆(fronto-temporal dementia)。Whyte等发现两例儿童型Paget骨病患者均存在该基因的纯合子缺失(断裂点相同,约缺失100kb)。患者血清中的OPG极低,而可溶性破骨细胞分化因子(sODF)明显升高。因而至少一部分儿童型Paget骨病与TNFRSFⅡB的纯合子缺失导致OPG缺乏有关。青少年Paget骨病常伴有进展性视网膜病。
25%~30%的家族性和部分散发性PDB与sequestosome基因1(编码NF-κB途径中的支架蛋白)突变(如P392L)有关;有人认为,sequestosome 基因1突变可能只是成为PDB的一个易感因素而非直接病因。患者常伴有慢性副黏病毒感染(para myxoviral infection),表达麻疹病毒核壳基因(measles virus nucleocapsid gene)的转基因小鼠发生PDB样骨损害。
VCP突变导致Paget骨病-肌病-痴呆三联征
SQSTM1基因突变是家族性PDB的常见病因,而散发性PDB的病因未明,涉及的遗传因素可能很多。近年发现,常染色体显性遗传性包涵体肌病(hereditary inclusion body myopathy)-Paget骨病(PDB)-额颞痴呆(frontotemporal dementia,FTD)症(IBMPFD,MIM 167320)是一种少见的高外显率的多系统性疾病,与散发性PDB有密切联系。IBMPFD以骨病-肌病-痴呆三联征为特点,其主要临床特点是:①肌病:属于包涵体肌病的一种,肌肉病变引起近端和远端肌无力(90%),常最先波及肩部和髋腰部肌肉,患者的乏力和虚弱呈进行性加重,不能抬腿升臂,上楼或登梯困难,肌肉酸痛,血清肌酶活性显著升高;最后可因呼吸肌与心肌病变而致死。②Paget骨病的临床表现与一般PDB相似,但发病年龄早,长骨和颅骨畸形及骨折常见,ALP和其他骨代谢标志物明显升高。③额颞痴呆的进展较快,由于大脑的额颞叶变性,语言和行为障碍特别明显,而撰写、绘画、计算和记忆力相对完好。少数患者伴有心肌病、肝纤维化、感觉和运动神经病变等。
IBMPFD是由于含空泡素蛋白(valosin-containing protein gene,VCP,CDC48,p97)突变所致,VCP是细胞代谢(cell metabolism)、胞膜融合(membrane fusion)、核膜重建(nuclear envelope reconstruction)、内质网相关物质降解(endoplasmic reticulum-associated degradation)、有丝分裂后高尔基池组装(post-mitotic Golgi cisternae reassembly)、泛素-蛋白酶体降解(ubiquitin-proteasome degradation)等过程的重要调节因子。目前,VCP突变引起IBMPFD的病例报道已有近200例,突变位点约16个。
骨病-肌病-痴呆三联征是诊断IBMPFD的必要条件,但一些患者可能不典型,其中以肌病的发生率最高(80%~90%),其次为PDB(43%~51%)和痴呆(30%~40%)。
破骨细胞对RANKL和VD受体过敏感
Paget骨病的骨病理学特点是破骨细胞数目明显增多、活性增高。Kakita等发现,多核细胞具有Paget骨病破骨细胞的特征,表现为细胞成熟更快更多(高于正常10~100倍以上),细胞内核数目增多,抗酒石酸酸性磷酸酶表达显著升高等,电镜下可见呼吸道融合病毒核壳(nucleocupsid),但未发现胞核或胞质包涵体。Demulder等发现培养液中的CFUGM克隆生成明显增加。用高度纯化的生血前身细胞(CD34+细胞和CD34-细胞)共培养时,Paget骨病的破骨细胞前身细胞和CFU-GM克隆明显增多。由CFU-GM克隆衍生的破骨细胞所需要的1α,25-(OH)2D浓度仅为正常时的1/10,这说明Paget骨病患者的破骨细胞前身细胞对活性VD有过度反应。Menaa等进一步证明,Paget骨病患者的破骨细胞前身细胞由于RANKL的表达过多而使其分化增殖为成熟破骨细胞的潜能增高,即患者的破骨细胞对RANKL存在过度反应,而这种过度反应又与患者骨髓中的M-CSF、IL-6过高有关。
IL-6通过NF-κB信号途径导致破骨细胞的功能调节失常,因而可认为Paget骨病是一种与NF-κB信号调节失常有关的疾病。其过程大约是IL-6升高引起 RANKL分泌增加,从而导致破骨性谱系细胞(包括前身细胞和成熟破骨细胞)生成过多和破骨细胞活性增强。破骨细胞前身细胞对1,25-(OH)2D过敏感的原因不是这些细胞的维生素D受体(VDR)过多或VDR变异。用GST/VDR融合蛋白进一步发现,该类患者的破骨细胞前身细胞表达的TAFⅡ-20(TFⅡD家族成员中的一种)增多,其意义是:当TAFⅡ-20含量够多时,破骨细胞前身细胞的分化就不依赖于1,25-(OH)2D,所以在1,25-(OH)2D很低时,破骨细胞的生成仍是加速的。
