
生理情况下,由肾上腺皮质分泌的皮质醇(cortisol)是成骨性谱系细胞和破骨性谱系细胞分化和功能调节的必需激素。但是,超生理量的皮质醇及其类似物则对骨组织的发育、生长与代谢有明显不利影响。1932年,Cushing等在报道Cushing综合征时就对糖皮质激素(glucocorticoids,GC)所致的骨质疏松(glucocorticoid-induced osteoporosis,GIOP)有了详细描述。GIOP是临床上常见的一种继发性骨质疏松,随着糖皮质激素应用的日益广泛,GIOP越来越常见。近年来,人们在努力开发新的糖皮质激素类药物,其目的是进一步减少蛋白分解和骨质疏松的不良反应。泼尼松、泼尼松龙、甲泼尼龙和地塞米松仍是广泛应用的主要口服糖皮质激素制剂,必须加强这些药物所致GIOP的防治。
GIOP在药物导致的骨质疏松中最为常见。糖皮质激素被广泛用于慢性非感染性炎性疾病(包括结缔组织病)、过敏性疾病及器官移植,骨质疏松为其最严重的副作用之一,即使是生理剂量的糖皮质激素也可引起骨丢失,绝经后妇女及50岁以上男性为高危人群。其中以关节疾病的患者接受长期、持续糖皮质激素治疗的可能性最高。多项纵向研究显示,糖皮质激素在治疗数周后,其骨量开始流失,最初数月内的骨量丢失迅速,可达5%~15%/年,而长期接受糖皮质激素治疗(1年以上)的患者骨质疏松发生率高达30%~50%。
糖皮质激素通过多个途径引起骨丢失(下图):
- 对抗Wnt/β-catenin信号,激活糖原合酶激酶β3 (GSK-β3),抑制成骨细胞的分化,抑制成骨细胞增殖,同时促进成骨细胞凋亡;
- 活化CCAAT扩增结合蛋白家族核因子,激活过氧化物酶体活性增殖γ2(PPARγ2),减少骨髓间质细胞向成骨细胞转化,诱导骨髓间质细胞向脂肪细胞转化;
- 促进破骨细胞的聚集和分化,破骨细胞凋亡减少,并增强其骨吸收活性;
- 降低肠钙吸收,尿钙排泄增加,血清甲状旁腺激素升高,尿钙排泄增加,导致骨量丢失;
- 骨保护素(OPG)下降,RANKL活性增加;
- 抑制胰岛素样生长因子生成和成熟成骨细胞功能;
- 降低垂体促性腺激素水平,抑制肾上腺雄激素的合成。
糖皮质激素所致的骨质疏松发病机制

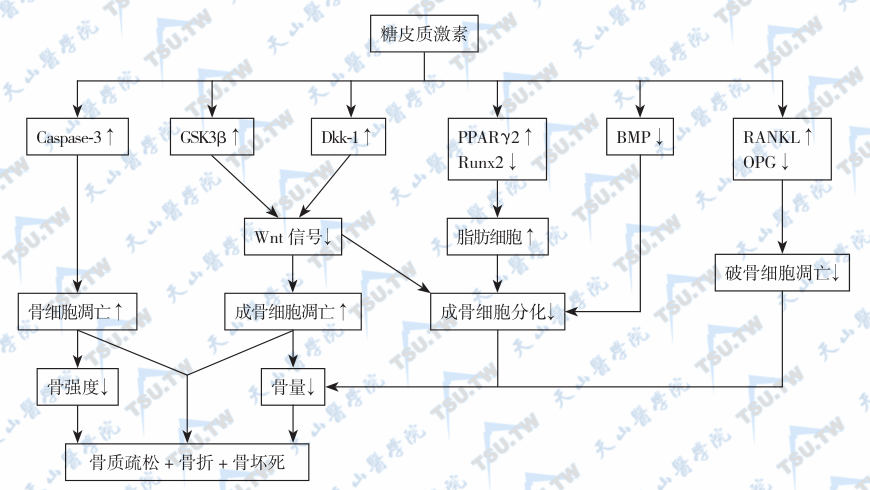
糖皮质激素所致的骨质疏松发病机制
注:Caspase-3:半胱天冬酶-3;BMP:bone morphogenetic protein,骨形态生成蛋白;Dkk-1:dickkopf-1;GSK3β:glycogen synthase kinase 3β,糖原合酶激酶3β;OPG:osteoprotegerin,护骨素;PPAR:peroxisome proliferator-activated receptor,过氧化物酶体活化受体;RANKL:receptor activator for nuclear factor-κB ligand,核因子-κB 配体活化素受体;Runx2:runtrelated protein 2,runt 相关蛋白2。
低骨量/骨质疏松是Cushing综合征的重要并发症
高皮质醇血症的病因有下列几种可能:
- Cushing综合征,如垂体ACTH瘤、垂体ACTH细胞癌、垂体ACTH细胞增生或肾上腺皮质肿瘤(分泌皮质醇的腺瘤或腺癌)、增生及原发性色素性结节性肾上腺结节等;
- 异源性CRH/ACTH分泌综合征;
- 糖皮质激素过敏感综合征,主要见于获得性免疫缺陷综合征(AIDS)患者。
糖皮质激素引起的BMD降低和骨质量下降呈糖皮质激素日剂量依赖性和糖皮质激素总剂量依赖性。糖皮质激素的用量在相当于泼尼松2.5~5.0mg/d即能增加骨折风险,而骨质量下降是骨折风险增加更重要的原因。
糖皮质激素引起骨吸收增强伴骨形成下降
高糖皮质激素血症引起骨丢失(糖皮质激素引起的骨质疏松,GIOP),其发病机制十分复杂,涉及许多激素、细胞因子和炎症因子的表达,如护骨素(osteoprotegerin,OPG)、RANKL (receptor activator for nuclear factor-κB ligand)、Runx2(runtrelated protein 2)、BMP、Dkk-1(dickkopf-1)、GSK3β(glycogen synthase kinase 3β)、PPAR(peroxisome proliferator-activated receptor)等。此外,糖皮质激素激活骨硬化素(osteosclerosis),使成骨细胞失活,从而抑制骨形成,而PTH和物理刺激(如负重)抑制骨硬化素表达,故可促进骨形成。
长期使用糖皮质激素后,松质骨出现过多脂肪细胞,成骨细胞缺乏伴骨吸收增强,骨小梁退化断裂;孔腔扩大、增多而缺乏破骨细胞,骨形成过程延长或缺陷。股骨头骨坏死区可见骨细胞和骨膜细胞大量凋亡。
GIOP的临床特征包括:
- 糖皮质激素制剂有一定差别;
- 接受治疗人群的敏感性有明显差别;
- 性别和年龄有明显差别;
- 使用途径和方法有一定差别;
- 剂量和疗程与骨丢失程度相关;
- 大剂量应用容易合并骨坏死。
但是,从临床防治的角度看,做到以下几点可显著降低GIOP风险:
- 尽量避免滥用糖皮质激素;
- 必须使用时选择最佳剂量、用法和疗程;
- 尽可能采用局部制剂;
- 隔日疗法保存下丘脑-垂体-肾上腺轴反馈功能;
- 病情控制即停用或使用最低有效剂量;
- 定期测量BMD,计划使用糖皮质激素时即采取措施预防;
- 具有循证依据的二膦酸盐为首选药物,普通VD有效,而活性VD对GIOP的治疗作用更强。
临床上常用的糖皮质激素类药物主要是泼尼松(强的松)、泼尼松龙(强的松龙)、甲泼尼龙和地塞米松。糖皮质激素通过其受体(GR、核受体和膜受体)而发挥作用。在体外实验中,糖皮质激素也可与盐皮质激素受体结合。成骨细胞可同时表达盐皮质激素受体(MR)、糖皮质激素受体GRα和GRαβ;破骨细胞可同时表达MR、GRβ和GRαβ;骨细胞可同时表达MR、GRα和GRαβ,但表达的量明显低于成骨细胞;肥厚性软骨细胞、增殖型软骨细胞和成熟软骨细胞也表达GRα、GRαβ和MR。这说明,骨组织细胞的功能均受糖皮质激素和盐皮质激素的双重影响。在糖皮质激素的作用下,骨重建每经历一次循环后,骨小梁的形成都被抑制而使骨小梁逐步变薄。过量的糖皮质激素抑制成骨细胞和破骨细胞蛋白、RNA和DNA合成。骨组织的分解代谢增强而合成代谢下降,导致骨量丢失和骨质疏松。
骨组织c-fos基因的启动子中含有糖皮质激素反应元件(GRE),糖皮质激素诱导成骨细胞c-fos、c-myc、骨涎蛋白(BSP)和骨连蛋白(osteonectin)基因表达。Id为细胞分化的抑制因子,地塞米松可使MC3T3-E1细胞的Id表达上调。糖皮质激素亦使成纤维细胞和成骨细胞Ⅰ型胶原α链、骨钙素、纤连蛋白、胶原酶、IGF-1、IGFBP-3、IGFBP-4、IGFBP-5和整合素β1、α2与α4表达下调,其最终后果是抑制成骨细胞的活性,促进破骨细胞生成和功能,故GIOP是一种以细胞因子表达紊乱为特征的代谢性骨病。
糖皮质激素通过促进破骨细胞介导的骨吸收及抑制成骨细胞介导的骨形成而引起骨质疏松,其作用机制包括:
- 影响钙稳态:糖皮质激素抑制小肠对钙、磷的吸收,增加尿钙排泄,引起继发性甲状旁腺功能亢进症,持续的甲状旁腺素(PTH)水平增高可促进骨吸收;
- 对性激素的作用:糖皮质激素可降低内源性垂体促性腺激素水平并抑制肾上腺雄激素合成,LH降低引起雌激素及睾酮合成减少;
- 抑制骨形成:长期应用糖皮质激素可抑制成骨细胞增殖,基质胶原和非胶原蛋白质的合成减少;
- 其他作用:糖皮质激素引起的肌病及肌力下降也可导致骨丢失。此外,患者本身的炎性疾病及合并用药(如环孢素)可导致骨质疏松。
骨形成和成骨细胞分化
在体内,糖皮质激素抑制骨形成;但在体外,糖皮质激素具有促进骨形成和成骨细胞分化作用,糖皮质激素可促进成骨细胞分化和矿化结节形成,但必须同时存在维生素C和β-甘油磷酸。低浓度糖皮质激素时促进矿化结节形成,但高浓度时无此作用。一般认为,生理浓度的糖皮质激素发挥的是一种允许作用(permissive role),它能促进骨生成,促进骨髓干细胞分化为成骨细胞,并且促进成骨细胞表型分子的表达,同时抑制单核细胞转化为破骨细胞。糖皮质激素亦促进骨髓基质细胞表达ALP、骨钙素、Ⅰ型胶原、骨桥素和骨涎蛋白。糖皮质激素对基质细胞上述作用的机制与BMP表达有关,BMP-2、BMP-4和BMP-6和糖皮质激素有协同作用。
过量糖皮质激素通过抑制成骨细胞的作用而诱发骨质疏松:①成骨细胞的生成减少,并通过MKP-1抑制成骨细胞增殖;②抑制成骨细胞前身细胞的分化和成熟成骨细胞,促进成骨细胞和骨细胞凋亡;③抑制Ⅰ型胶原合成;④血清骨钙素明显下降,昼夜节律性消失。
骨吸收和破骨细胞功能
药理浓度的糖皮质激素促进破骨细胞生成和RANKL表达,抑制OPG表达,RANKL/ OPG比值升高,血清OPG明显降低,故破骨细胞的骨吸收功能增强。糖皮质激素抑制成骨细胞IGF-1表达。应用糖皮质激素后,由于骨吸收增加,迅速出现骨量丢失(骨量丢失的急性相),在松质骨中的破骨细胞数目增多,寿命延长。但在体外实验中,地塞米松使破骨细胞前身细胞增殖和分化受抑,故在一般情况下,GIOP的破骨细胞活性不高。
糖皮质激素代谢
糖皮质激素受体数目与细胞中的11β-HSD2表达水平直接相关。因此,11β-HSD1和11β-HSD2调节了成骨细胞对糖皮质激素的敏感性。而GH、IGF-1、TNF-α等可抑制11β-HSD1活性,增强了骨组织对糖皮质激素的敏感性。
靶细胞的糖皮质激素由两种酶催化其代谢。在2型11β-羟甾体脱氢酶(11β-HSD2)的催化下,糖皮质激素转化为无生物活性的糖皮质激素代谢产物(单向催化);1型11β-HSD(11β-HSD1)为一种双向催化酶,可使地塞米松与11-脱氢地塞米松之间的反应双向进行,而两种形式的地塞米松均有生物活性。11β-HSD1具有氧化还原酶特点,因此在一定条件下,使无活性的糖皮质激素重新获得生物活性。11β-HSD2主要在肾脏和胎盘中表达,使盐皮质激素受体不被糖皮质激素激活。在组织中,皮质醇/促皮质素(cortisone)通过上述的两种11β-HSD的催化作用,进行着循环性的双向反应。由于GH和IGF-1对11β-HSD1表达有抑制作用,当GH和(或)IGF-1缺乏(如GIOP)时,肾、肝、脂肪细胞及骨骼中的内源性皮质醇生成增多。
维生素D代谢
长期使用糖皮质激素后,24-羟化酶表达上调,同时1α-羟化酶表达下调,血1,25(OH)2D减低,这可能是患者肠钙吸收减低,尿钙排出增加的重要原因。
骨坏死
糖皮质激素引起的骨坏死表现为无血管性骨坏死或无菌性骨坏死,为长期应用糖皮质激素或多次使用特大剂量的糖皮质激素的重要并发症之一,占全部骨坏死病例的16%~34%;股骨头、肱骨头和肱骨远端为好发部位,但也可见于四肢的其他长骨。骨坏死发生率和严重程度与糖皮质激素的疗效和剂量有关,但短期应用或关节内应用也可发生。骨坏死的发生机制未明,有如下几种可能:
- 糖皮质激素使骨骺变性,骨疲劳损伤致骨小梁微破裂(microcracks);
- 缺血性坏死可能与脂肪栓栓塞血管有关;
- 骨内脂肪堆积,骨内压升高(Cushing综合征的表现之一),血管床损伤,血流减少;
- 股骨颈的成骨细胞和骨细胞凋亡增加。
在动物实验中,GIOP家兔的骨小梁骨损害严重,在激光显微镜下可见新生的编织骨吸附四环素荧光。新生的编织骨深入骨小梁内部。关节骨膜下的骨小梁基质明显受损(与成骨细胞和骨细胞凋亡有关),护骨素/破骨细胞形成抑制因子(OPG/OCIF)的表达明显降低。
糖皮质激素间接导致骨丢失
GH
泼尼松可抑制垂体GH分泌。用生物分析测定IGF-1的活性发现,IGF-1的促细胞生长发育作用下降,可能是因为长期应用糖皮质激素后,血清中出现了IGF-1的抑制物,这些抑制物很可能就是IGFBPs(如IGFBP-1)。
LH/FSH
在糖皮质激素的作用下,垂体分泌的LH 和FSH减少,分泌反应迟钝。GnRH细胞的糖皮质激素受体与地塞米松结合,抑制其合成和分泌。此外,糖皮质激素对卵巢、睾丸和肾上腺的性腺甾体类激素合成也有抑制作用,因此长期应用糖皮质激素者(女性)的血清雌二醇、雌酮、DHEA、雄烯二酮和孕酮均下降(男性患者则有DHEA和睾酮下降),腰椎BMD与血清雌二醇呈正相关,同时肾上腺皮质的雄激素合成被抑制,血清DHEAS和雄烯二酮明显下降。
PTH和维生素D
长期应用糖皮质激素治疗的患者胃肠钙的吸收障碍,出现高钙血症和高尿钙症,血钙、尿钙与血1,25-(OH)2D呈正相关;肾小管磷重吸收率下降,负钙平衡及继发性甲旁亢。糖皮质激素直接抑制Na+依赖性磷的重吸收。在糖皮质激素的作用下,成骨细胞对PTH的敏感性增高。
其他因素
糖皮质激素增加尿钙排出,抑制肠道吸收,增加蛋白质分解,严重时引起肌肉消耗和糖皮质激素所致的肌病(glucocorticoid-induced myopathy),这些因素均可引起骨盐丢失和骨质疏松。
到目前为止,GIOP的病因与发病机制可总结为如下6点:
- 直接损害成骨细胞、骨细胞和破骨细胞功能,骨形成减少,骨吸收增多;但与一般骨质疏松不同的是GIOP以骨形成缺陷为主;
- 骨重建功能减退,骨微损伤后修复能力下降,骨脆性增加,易发生骨折和骨坏死;
- PTH分泌过多,引起继发性甲旁亢;
- 糖皮质激素直接或通过间接途径(如非昼夜节律性作用)拮抗性腺功能,抑制性腺激素、GH和IGF-1的骨形成作用;
- 糖皮质激素引起肌肉萎缩和肌无力,骨骼的应力负荷降低;
- 肠吸收和肾小管重吸收钙减少,负钙平衡促进继发性甲旁亢的进一步发展。
