
目前关于恶液质的具体机制尚不清楚,以下几个方面与恶液质密切相关:①机械性因素,如消化道肿瘤梗阻等;②炎症因子紊乱;③机体代谢,特别是蛋白质代谢失调;④厌食与促食因子(anorexigenic and orexigenic factors)失衡。疾病与宿主之间的相互作用引起的一系列变化可以通过mTOR通路促进恶液质的发生发展。
mTOR与肿瘤性恶液质
Schmitt TL等在16例怀疑有肿瘤而行胰腺切除的患者(术前根据体重下降情况均分为恶液质组和非恶液质组)的肝脏和肌肉活检标本中进行蛋白检测,发现骨骼肌中的mTOR和p70S6K磷酸化水平明显降低,且体重下降越明显磷酸化水平越低;肝脏中的PI3K/Akt/mTOR通路反而被激活,提示mTOR、p70S6K与恶液质发生密切相关。虽然纳入病例较少,但是仍然有重要的提示意义。这个结果与临床上观察到的肿瘤恶液质患者外周循环中急性期蛋白升高的现象是一致的,即肿瘤条件下,肌组织蛋白被消耗,内脏蛋白的合成反而增加。Robert F等通过敲掉淋巴瘤小鼠模型(Eµ-Myc)中mTOR通路上重要负向调节因子PTEN、TSC1或者TSC2,成功建立了荷TSC2+/-Eµ-Myc淋巴瘤小鼠的肿瘤恶液质模型。与单纯Eµ-Myc小鼠相比,TSC2+/-Eµ-Myc小鼠表现出一系列炎症因子和细胞因子的失调,包括IL-10增加,瘦素和神经肽Y(neuropeptide Y,NPY)下降等。肿瘤细胞移植后,小鼠在9~11天出现体重下降,骨骼肌和瘦体组织降低,食欲减退等典型恶液质表现,生存预后也更差。然而当给予mTOR抑制剂雷帕霉素或者高三尖杉酯碱(homoharringtonin)后,TSC2+/-Eµ-Myc小鼠的IL-10水平下降,恶液质状态和预后均得到改善。C26结肠癌动物恶液质模型中,IL-6的水平同样也受到雷帕霉素和高三尖杉酯碱的调节。提示mTOR可能是这些恶液质相关细胞因子下游作用靶点,抑制蛋白质合成可以改善恶液质状态,具体机制尚有待进一步研究。White JP等通过导入外源性IL-6表达载体,在Apcmin/+小鼠成功诱导恶液质表型,且随着IL-6水平升高,骨骼肌中mTORC1及其下游分子磷酸化水平亦进行性降低,进一步证实了肿瘤恶液质中失调的细胞因子可以通过mTOR通路降低蛋白合成水平,引起体重下降。同样是在Apcmin/+肿瘤恶液质模型中,小鼠心肌也表现出消耗现象。但有趣的是,不仅其消耗程度不及骨骼肌,机制上也有所不同。在心肌上,AMPK/ mTOR 通路起主要作用,而与Akt无关。
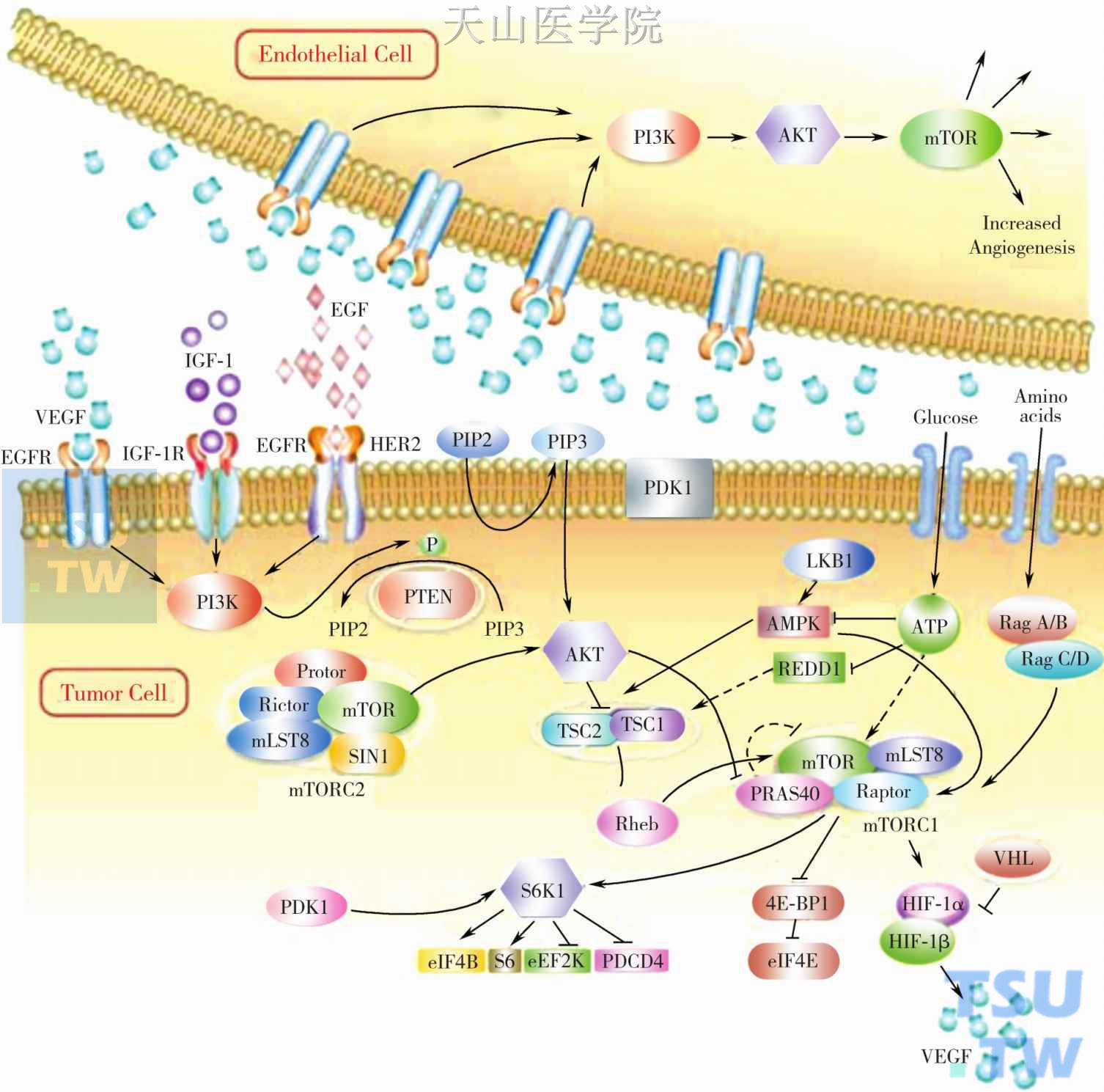
图2-9-2 mTOR的信号传导通路
PDGF,platelet derived growth factor,血小板衍生生长因子;BDNF,brain derived neurotrophic factor,脑源性神经营养因子;EGF,epidermal growth factor,表皮生长因子;FGF,fibroblast growth factor,成纤维细胞生长因子;NGF,nerve growth factor,神经生长因子;MAPK,mitogen activated protein kinase,丝裂源活化蛋白激酶;ERK,extracellular signal-regulated kinase,细胞外信号调节激酶
VEGF
VEGFR EGFR HER2 PIP2 PIP3
骨骼肌约占正常成人体重的40%,是瘦体组织的主要成分,也是恶液质状态下的主要蛋白丢失场所。在MAC16荷瘤小鼠恶液质模型中,腓肠肌4E-BP1和p70S6K1的磷酸化水平明显下降,使得起始复合体形成减少,导致小鼠体重丢失。而支链氨基酸(branched chain amino acids,BCAAs),特别是亮氨酸能够部分逆转恶液质小鼠的症状。机制是亮氨酸及其诱导分泌的胰岛素可以激活mTOR/4E-BP1和p70S6K1通路,启动蛋白质合成。胰岛素/mTOR还可以促进eEF2去磷酸化,解除其对蛋白合成延长的抑制,起到拮抗恶液质的作用。也有证据表明除了前面所述机制外,mTOR还可以被AMPK激活,参与必需氨基酸诱导的蛋白合成,这与White JP等的研究结果是一致的。有些学者也报道了胰岛素/mTOR的激活可以抑制UPS中E3连接酶Atrogin-1和MuRF-1的表达,从而下调骨骼肌蛋白的降解,转录因子Foxo是其中的关键蛋白(图2-9-3)。
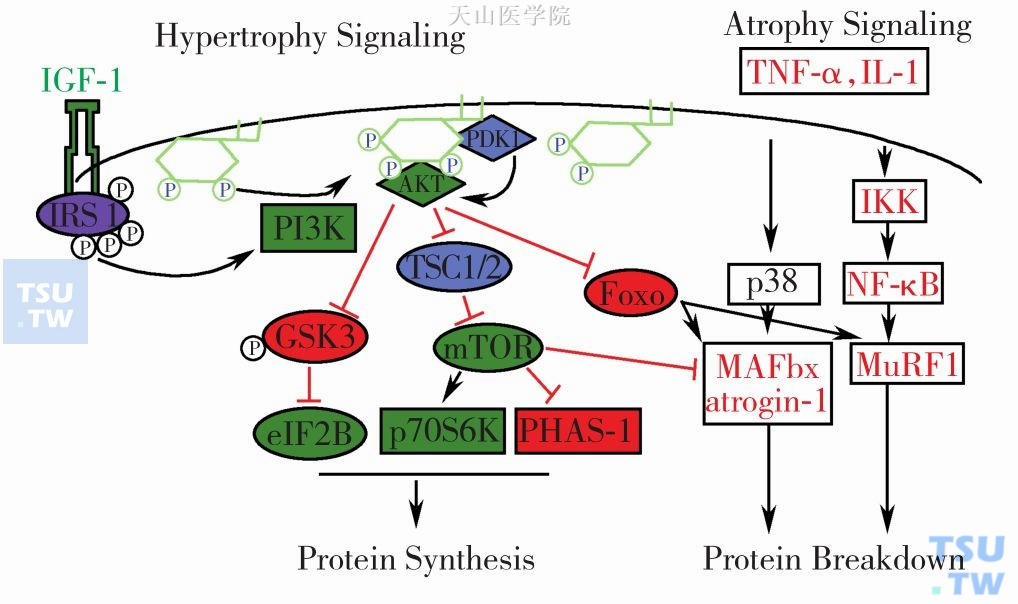
图2-9-3 mTOR调节骨骼肌肥大与萎缩
mTOR除了促进蛋白合成之外,也与瘦素介导的厌食症相关。Cota D等研究发现,磷酸化mTOR以及S6K1主要存在于下丘脑的室旁核(paraventricular,PVN)和弓状核(arcuate nuclei,ARC)区域中,而这里正是摄食中枢。当在大鼠侧脑室给予瘦素注射后,下丘脑中的mTOR通路被激活,动物进食明显减少,并伴有体重下降。亮氨酸可以通过相似的机制导致厌食症的发生,而雷帕霉素则能阻断上述效应。说明mTOR与瘦素介导的厌食症有一定相关性(图2-9-4)。
mTOR与心源性恶液质
心源性恶液质是指心力衰竭时伴发的肌肉组织丢失,其往往是预后不良的征兆。AngⅡ在心力衰竭发病中起着重要作用,伴发的恶液质状态下其作用同样受到广泛关注。AngⅡ持续灌注可以诱导小鼠发生心力衰竭-恶液质综合征,且模型动物表现出与恶液质患者相似的细胞因子紊乱。循环和骨骼肌中IGF-1水平下降,但是IGF-1静脉注射或者肝脏特异性IGF-1基因无效突变并不能阻止小鼠体重丢失。只有在肌肉中特异性高表达IGF-1时才可以阻断AngⅡ的心力衰竭恶液质诱导效应。此时骨骼肌mTOR及下游分子磷酸化水平增加且同时伴有Atrogin-1和MuRF-1表达水平下降,提示IGF-1可以通过mTOR通路促进蛋白合成和抑制蛋白分解。
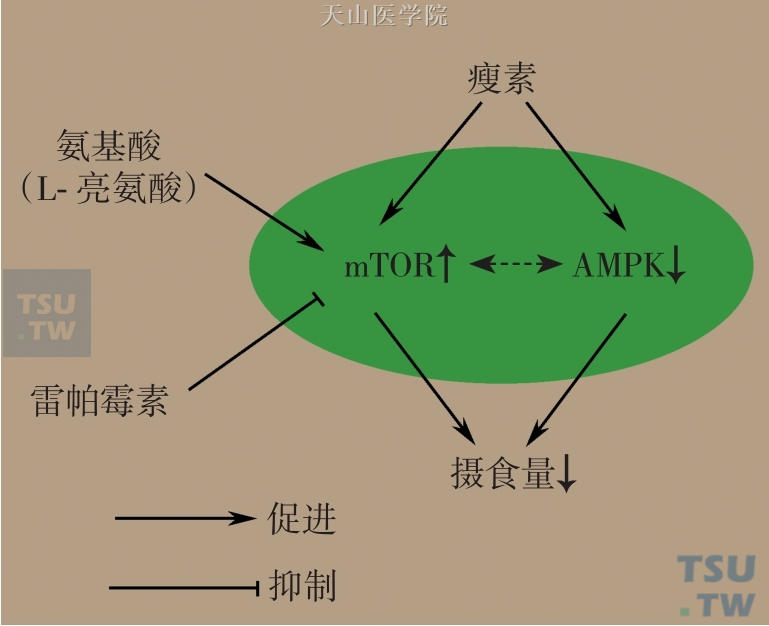
图2-9-4 下丘脑mTOR与厌食发生的关系
mTOR与重症恶液质
在急危重症(外伤、手术并发症、严重感染等)中,往往伴发体重下降和骨骼肌消耗,这种疾病状态下恶液质的形成机制非常复杂。Constantin D等通过对健康志愿者和各种原因所致的危重患者(不包括神经肌肉性疾病、慢性阻塞性肺疾病、充血性心力衰竭)检测发现,外周血中TNF-α和IL-6明显升高,而肌组织活检提示mTOR及其下游分子的磷酸化水平明显降低,提示mTOR与危重患者蛋白消耗有关。
mTOR与失用性萎缩
由于骨折、制动、卧床以及机械通气等所造成的失用性萎缩中,主要原因是氨基酸吸收后水平蛋白的合成减少。动物实验证实了myostatin、p38MAPK、促炎因子可以导致胰岛素和IGF-1抵抗,从而可以通过IGF-1/Akt/mTOR通路抑制蛋白质合成。也有实验表明在失用性萎缩大鼠模型中,mTORC1抑制剂REDD1/2表达上调,同时其下游p70S6k的信号传导降低。
mTOR与肾源性恶液质
各种原因所致的慢性肾病终末期所出现的骨骼肌、脂肪组织减少,体重显著下降,食欲减退,厌食,贫血等称为尿毒症性营养不良-恶液质综合征。终末期肾病患者伴随的代谢性酸中毒、循环毒素、系统性炎症以及透析等治疗措施均可引起胰岛素和IGF-1抵抗,通过Akt/mTOR通路导致蛋白合成减少。
mTOR与肌肉减少症
肌肉减少症是指随着年龄的增加,出现肌肉萎缩,并同时伴有骨骼肌力量的下降。目前对于蛋白质合成减慢在其发病中的作用,不同研究得出不同的结论,但是在动物模型或者老年人中,骨骼肌对外界合成代谢的刺激不敏感是为大家所公认的。即随着年龄的增加,激活PI3K/Akt/mTORC1通路所需的胰岛素的剂量也增加,表明该通路与肌肉减少症患者“合成代谢抵抗”相关。
总之,各种原因所致的恶液质均表现出体重下降和肌组织消耗,而mTOR因其促进蛋白合成和抑制蛋白分解的功能,与恶液质的发生密切相关。
