
脂肪不断分解消耗是恶液质的重要特征之一。这表明恶液质患者脂类代谢出现严重的紊乱。许多研究认为脂代谢改变可能与人类和动物各种肿瘤的发展和癌症患者的恶液质密切相关,脂代谢改变可能是恶液质的一个重要致病因素。因此,提出抑制脂类分解可抑制肿瘤生长。恶液质患者脂类代谢主要表现为:脂动员增加,内源性脂肪消耗增加,脂肪储备减少,且不受葡萄糖输入抑制影响;脂肪酸氧化分解增加;血浆脂蛋白(CM和VLDL)升高,血浆甘油三酯水平升高,外源性脂肪利用下降。
关于恶液质患者脂代谢紊乱机制还没有完全阐明,基本上与肿瘤和宿主释放的一些因子,以及激素紊乱等密切相关。这些因子和激素通过相关信号通路激活HSL激活和抑制脂蛋白脂酶(LPL),最终导致恶液质患者脂肪不断消耗和高脂血症。许多研究发现即使非侵袭性肿瘤并且没有发生营养摄入改变的肿瘤患者,都显示腹膜后储存脂肪的严重下降,这提示肿瘤或宿主体内产生了一种分解脂类的物质并释放入血液中。这种脂代谢紊乱和循环中脂解活性因子在癌症早期就存在,并且随着癌症进展而愈来愈严重。如卵巢癌患者血清和腹腔液中可检出促进脂类分解的活性物质,激素敏感脂肪酶(HSL)活性是正常人的2.3倍。同时发现患者腹腔液中具有诱导HSL表达的活性因子。ATGL也与HSL一样在恶液质脂肪消耗中发挥重要作用,最新动物模型研究发现ATGL缺失的荷瘤鼠不会出现WAT消耗,同时也不出现骨骼肌降解,这表明ATGL和HSL一起参与了肿瘤恶液质的脂肪消耗。
LMF/ZAG
许多研究证实脂肪动员因子(lipid mobilizing factor,LMF) 为一种分子量45kD,并且可被胰酶水解为小分子后仍具有活性,具有耐热和耐水解的蛋白质。将该分子注入小鼠体内后立即产生脂肪动员、厌食和恶液质。根据氨基酸序列分析和免疫学性质发现LMF与一种已知锌-α2-糖蛋白(zinc α2 glycoprotein,ZAG)为同一种物质,具有促进机体脂肪分解和增加产热双重途径减少体内脂肪,肿瘤组织可分泌ZAG,而脂肪细胞分泌的ZAG可能通过自分泌或旁分泌的方式作用于局部脂肪组织,从而使脂肪分解。可表达ZAG的正常组织包括肺、心、BAT 和所有白色脂肪组织(WAT),而脂肪细胞不仅表达而且可分泌ZAG。在脂肪组织下降60%的荷瘤鼠模型上,WAT的ZAG mRNA表达水平增加10倍,BAT增加3倍,而同时发现WAT的瘦素(Leptin)下降了33倍,脂联素没有变化,而当体重下降达24%时,ZAG 蛋白质水平分别在WAT和BAT中下降了10倍和20倍。这表明ZAG是一个重要脂肪细胞因子,并在脂肪代谢和恶液质发生过程中发挥着重要作用。在LMF/ZAG主要通过经典依赖GTP的腺苷酸环化酶-cAMP通路激活HSL(图3-3-3)。动物模型研究表明LMF/ZAG能够消除过度喂养鼠40%脂肪,以及ob/ob鼠19%脂肪,而并不影响动物体液和非脂肪组织重量,同时人体内也发现类似作用。LMF/ ZAG促进脂肪动员同时还加强脂类的氧化分解,其机制可能通过激活肾上腺能受体β3-腺苷酸环化酶-cAMP通路而促进UCP1表达有关;同时通过这条通路还能促进UCP2表达,而这与消除自由基毒性和抵抗化疗作用有关。LMF/ZAG还以剂量依赖方式并通过MAPK通路促进UCP3表达。小鼠模型研究发现LMF/ZAG还增加脑、心、BAT和骨骼肌利用葡萄糖和降低血糖,同时以依赖GTP方式激活腺苷酸环化酶而消耗肝糖原。多种机制可以调节ZAG的表达:人脂肪细胞研究发现PPARγ激动剂罗格列酮可以诱导ZAG表达上调达3倍,TNF-α可上调ZAG4倍,同时ZAG还受到肾上腺能受体β3激动剂BRL37344和糖皮质激素调节。而许多动物实验和临床研究提示糖皮质激素可能是恶液质ZAG表达升高主要调节因素:糖皮质激素拮抗剂RU38486可以明显减轻恶液质的体质下降和WAT的ZAG水平;皮质醇增强是恶液质早期的一个特症,恶液质鼠血浆皮质醇水平与体重丢失成正比,营养不良的恶液质患者尿液中皮质醇增强的,还有儿茶酚胺类增加也与恶液质ZAG水平有关。二十碳五烯酸ω-3脂肪酸如(eicosapentaenoic acid,EPA)对恶液质有一定疗效,细胞实验发现EPA可以减轻由地塞米松诱导的人脂肪细胞的脂肪降解和ZAG表达,这可能EPA治疗恶液质的一种机制(图3-3-4)。
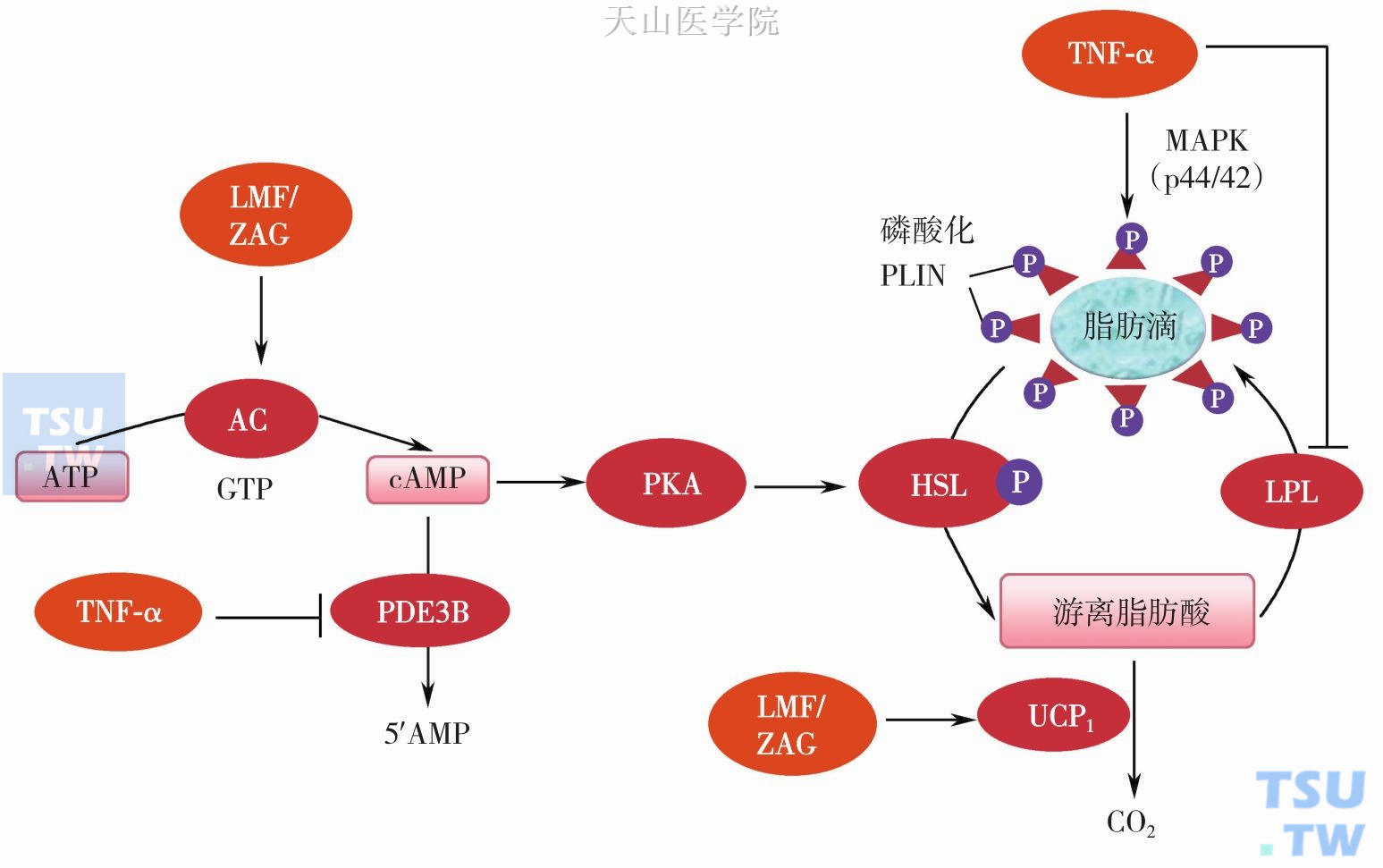
图3-3-3 LMF/ZAG和TNF-α调节脂肪细胞甘油三酯的机制
LMF/ZAG,脂肪动员因子/锌-α2-糖蛋白;TNF-α,肿瘤坏死因子-α;PKA,蛋白激酶A;HSL,激素敏感脂肪酶;PDE3B,磷酸二酯酶3B;UCP1,解偶联蛋白1,M APK,丝裂原激活蛋白激酶;PLIN,周脂素
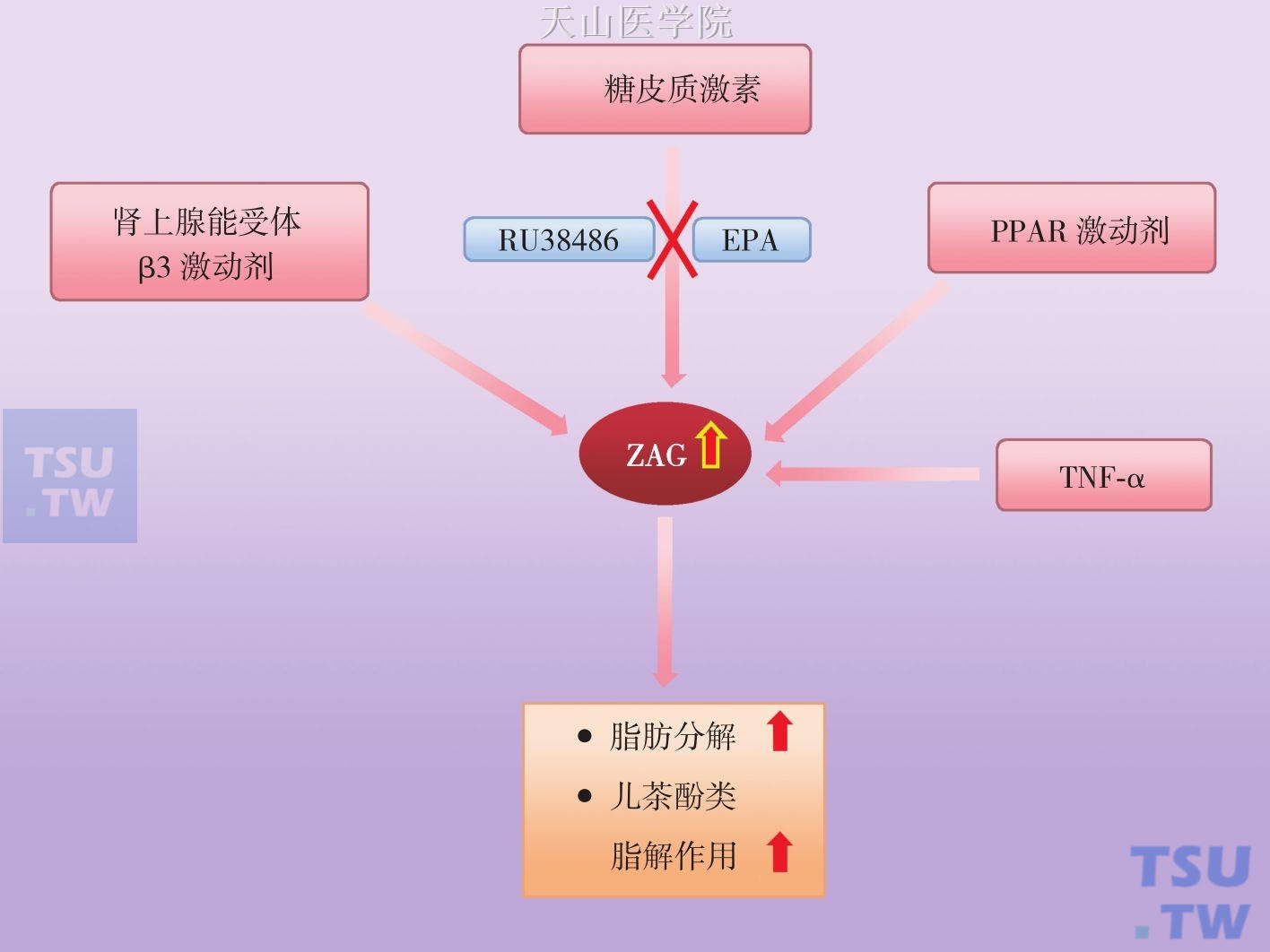
图3-3-4 影响白色脂肪组织ZAG表达的因素
TNF-α,肿瘤坏死因子-α;EPA,二十碳五烯酸;RU38486,糖皮质激素受体拮抗剂
TNF-α
相当多的研究发现TNF-α肿瘤恶液质患者脂肪消耗的主要细胞因子之一。TNF-α可以通过抑制脂蛋白脂酶(LPL)表达而限制外源性脂类摄取,或通过直接促进脂肪分解等途径来降低恶液质患者的脂肪(图3-3-3)。TNF-α促进脂肪分解机制涉及MAPK p44/42和JNK(c-jun-NH2-terminal kinase)通路激活,主要通过专一性的抑制水解cAMP的磷酸二酯酶3B而升高细胞内cAMP水平。MAPK通路激活后,一方面可以抑制PLIN表达,同时磷酸修饰PLIN使之脱离脂肪滴表面,促进脂肪分解;TNF-α还可以激活NFκB通路促进脂肪水解;MAPK p44/42和JNK通路激活后磷酸化PPARγ,从而阻止PPARγ的转录激活功能。另外,与LMF/ZAG一样,TNF-α可以明显诱导骨骼肌UCP2和UCP3的表达而促进产热作用。
IL-1、IL-6和IFN-γ
TNF-α可以诱导IL-6合成,并一起作用:一起激活炎症级联反应,促进急性期反应(APR)导致机体高分解代谢。机体APR程度与血清TNF-α和IL-6水平呈明显正相关。结肠腺癌小鼠模型发现IL-6水平与恶液质发展相关并且用IL-6抗体治疗能明显改善体重下降和其他恶液质指标。IFN-γ抗体治疗在动物模型上也有明显效果。然而一组61例癌症晚期患者研究发现TNF-α,IL-1、IL-6和IFN-γ血清水平与体重下降没有明显。与TNF-α一样,IL-1、IL-6和IFN-γ都能抑制LPL表达,同时IL-1和IFN-γ也能直接激活脂肪降解而IL-6没有。
