
嗜铬细胞瘤(pheochromocytoma)是起源于神经嵴的嗜铬细胞肿瘤,肿瘤细胞主要合成和分泌儿茶酚胺(catecholamine,CA),故又称为儿茶酚胺分泌瘤。这些肿瘤的绝大多数来源于肾上腺髓质的嗜铬细胞,即通常意义上的“嗜铬细胞瘤”。来源于肾上腺外嗜铬组织的肿瘤可分为神经节神经瘤、副神经节瘤、化学感受器瘤和颈动脉体瘤等,可统称为肾上腺外嗜铬细胞瘤(extra-adrenal pheochromocytoma)。因副神经节属于特殊分化了的神经嵴细胞,这些细胞在移行过程中可散布于机体的任何部位,故肾上腺外嗜铬细胞瘤可发生于任何器官,但是大多数位于从颈部至膀胱的交感神经节或颈动脉体、迷走神经体(vagal body)、纵隔、主动脉或盆腔Zuckerkandl器。嗜铬细胞瘤可为遗传性,并作为多发性内分泌腺瘤综合征(MEN-2A、MEN-2B、von Hippel-Lindau病或多发性神经纤维瘤病Ⅰ型等)的一部分。偶尔,肾上腺或肾上腺外组织可发生嗜铬细胞瘤-神经节神经瘤(pheochromocytoma-ganglioneuroma)。瘤细胞阵发性或持续性分泌大量儿茶酚胺,临床上表现为阵发性或持续性高血压及代谢紊乱综合征;某些肿瘤(如化学感受器瘤)可能不具备儿茶酚胺产生和分泌能力,称为球瘤(glomoma),多位于颈动脉体、颈静脉窦及中耳等处。副神经节遍布腹部和盆腔神经丛,但一般均在出生后2~3年内消退。如退化不良,可形成增生结节或嗜铬细胞瘤。散发型嗜铬细胞瘤常为单个,80%~85%的肿瘤位于肾上腺内,右侧略多于左侧,少部分位于肾上腺外嗜铬组织。儿童患者的肾上腺外和双侧肾上腺嗜铬细胞瘤发病率较成年人高。
随着研究的深入,它已不能表达其临床表现和病理生理本质,不少病例是由于嗜铬细胞增生引起的,并未形成肿瘤;虽然“嗜铬细胞瘤”以分泌过量儿茶酚胺为特征,但还同时伴有神经肽及APUD激素的分泌亢进。所以,“嗜铬细胞瘤”的命名并不确切。
嗜铬细胞瘤的发病率较低,在初诊的高血压患者中占0.1%~0.5%。各年龄段均可发病,发病高峰30~50岁,男女发病率基本相同。80%~90%的嗜铬细胞瘤为良性,恶性占10%~16%。部分患者在生前即得到确诊,而另一些患者在尸解时才被发现。嗜铬细胞瘤可造成心、脑、肾等重要脏器的严重损害,甚至危及生命。如能早期诊断,绝大多数手术切除后治愈。
散发性嗜铬细胞瘤与交感神经副神经节瘤的病因未明。目前的病因研究主要集中在遗传性嗜铬细胞瘤与交感神经副神经节瘤方面。
RET/MEN-1/VHL/SDH突变导致遗传性嗜铬细胞瘤/副神经节瘤
大约24%的嗜铬细胞瘤与交感神经副神经节瘤为家族性肿瘤综合征(familial cancer syndrome)的组成部分,表现为MEN-2、家族性副神经节瘤、von Hippel-Lindau病、1型神经纤维瘤病或嗜铬细胞瘤-副神经节瘤综合征(pheochromocytoma-paragenelioma syndrome)等5种类型。家族型嗜铬细胞瘤多位于肾上腺内,常为多发性,累及双侧肾上腺,肾上腺外少见。因此,对有家族史的患者及其一级亲属应进行常规遗传学筛查。15%~20%的家族型嗜铬细胞瘤是由于种系细胞突变(胚系突变,germline mutation)所致,突变的基因存在于患者体内的所有细胞中,这些患者的共同特点是年龄小,发病呈家族聚集。25%~35%的副神经节瘤亦为家族性,根据琥珀酸脱氢酶亚基突变的区别可分为若干种亚型(下表)。
副神经节瘤的遗传学分型
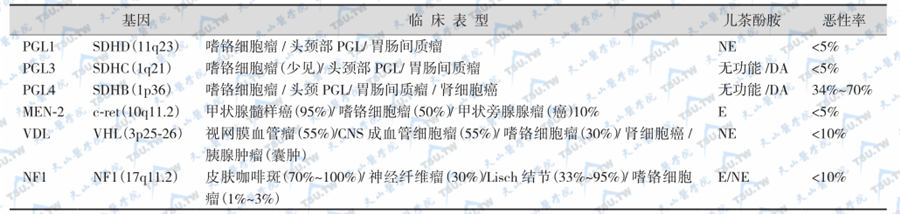
注:PGL:paraganglioma,副神经节瘤:SDH:succinate dehydrogenase,琥珀酸脱氢酶;VDL:von Hippel-Lindau瘤;NF1:神经纤维瘤病1型;NE:去甲肾上腺素;E:肾上腺素;DA:多巴胺
RET突变
1993年和1994年,分别在多发性内分泌腺瘤病2A(MEN-2A)与(MEN-2B)中发现了RET种系突变。MEN-2A(Sipple综合征)为常染色体显性遗传病,外显率主要与年龄有关。表现为肾上腺嗜铬细胞瘤(绝大多数为双侧性)、甲状腺髓样癌及甲状旁腺腺瘤引起的甲旁亢。Felix Fränkel 于1886年首次报道的嗜铬细胞瘤病例为18岁女性,双侧肾上腺存在“肉瘤和血管肉瘤”,有嗜铬细胞瘤的典型临床表现。120年后(2007年)检查其后代,发现4位亲属有RET基因的种系突变。因此,1886年首次报道的嗜铬细胞瘤病例的诊断实际上是MEN2B。MEN-2B患者除肾上腺嗜铬细胞瘤(双侧性)、甲状腺髓样癌及甲旁亢外,还有黏膜神经瘤、角膜神经增厚、肠神经节神经瘤病(intestinal ganglioneuromatosis)和Marfan综合征样体形等。
MEN-2伴嗜铬细胞瘤具有如下特点:①嗜铬细胞瘤为双侧性,但发病时间可相差悬殊;②因差异性基因表达关系,肿瘤主要合成和分泌肾上腺素和甲氧肾上腺素(metanephrine),故患者的心动过速和血糖升高更为突出;③酪氨酸羟化酶活性增强,儿茶酚胺及其代谢物水平较高,症状较为明显。40%~50%的MEN-2A可发生嗜铬细胞瘤,常为多发性,双侧性;肿瘤周围可有弥漫性或结节性增生,主要分泌肾上腺素,故早期仅有血液或尿生化的改变。
MEN-1突变
MEN-1为肿瘤抑制基因MEN-1失活性突变所致,呈常染色体显性遗传,其临床特点是垂体瘤、原发性甲旁亢、胰岛细胞瘤、血管纤维瘤和脂肪瘤,偶尔合并嗜铬细胞瘤,主要分泌去甲肾上腺素和甲氧去甲肾上腺素(normetanephrine)。
VHL突变
与RET基因相反,VHL是一种抑癌基因,编码两种蛋白。目前已发现200多种突变类型,突变基因所表达的蛋白有丧失功能和获得功能两种。70%~90%的患者为种系突变,其他为体细胞突变;前者决定VHL家族的肿瘤易感素质及发病情况,而后者与肿瘤的恶性倾向有关。von Hippel-Lindau综合征由嗜铬细胞瘤(10%~90%)、视网膜血管瘤、中枢神经成血管细胞瘤、肾癌、肾脏和胰腺囊肿及多发囊腺瘤组成。VHL综合征可分为1型和2型两种,2型VHL可再分为A、B、C 3种亚型,各型表型的主要差别见下表。
2型VHL综合征的亚型表型

患者有阳性家族史和一种典型肿瘤(视网膜、脊髓、小脑成血管细胞瘤,肾癌或嗜铬细胞瘤),或虽然无阳性家族史,但有两个部位以上的成血管细胞瘤,或有一个部位的成血管细胞瘤但有肾癌或嗜铬细胞瘤。确诊可针对患者及亲属作基因分析以证明VHL突变,并用转染技术证实突变VHL蛋白的功能。
SDH突变
线粒体琥珀酸脱氢酶(succinate dehydrogenase,SDH)是三羧酸循环和有氧电子传递呼吸链中的关键酶之一,包含A、B、C、D4个亚基。亚基A(SDHA,黄素蛋白)和B(SDHB,硫铁蛋白)形成酶接触中心,而亚基C (SDHC)和D(SDHD)组成酶的锚定区。4个亚基分别由4个基因编码。SDHA基因编码黄素蛋白,SDHB编码硫铁蛋白亚基,SDHC基因编码琥珀酸-辅酶Q氧化还原酶中细胞色素b大亚基(cybL),而SDHD编码琥珀酸-辅酶Q氧化还原酶中细胞色素b小亚基(cybS)。
在家族性和散发性嗜铬细胞瘤中存在SDHD、SDHB基因突变。作为SDH的亚基,SDHD锚定区和SDHB的酶接触中心对维持线粒体SDH的活性是必需的,SDHD或SDHB基因突变可使线粒体氧传感通路异常,造成组织缺氧,进而在低氧诱导因子-1(HIF-1)的诱导下促进VEGF及其受体(VEGFR)基因转录,参与肿瘤的血管形成。在散发性嗜铬细胞瘤中检测到的SDHD突变有G14A、C33A、36/37delTG、52+2(IVS1+2)T、C112T、G274T和C361T等,其中6个突变可导致蛋白质合成提前终止,1个突变可致氨基酸替换;SDHB基因突变有9个。据报道,50%的肾上腺外嗜铬细胞瘤有SDHB基因突变,36%有SDHD基因突变。
NF1突变
嗜铬细胞瘤只与Ⅰ型多发性神经纤维瘤病有关,其基本病因为NF1基因失活。该基因失去表达则导致嗜铬细胞瘤及其他肿瘤,其中的嗜铬细胞瘤多为单侧良性,偶为双侧甚至伴有腹部副神经节瘤。
其他变异
神经-皮肤综合征中的副神经节瘤有强烈的遗传背景,但具体病因未明。分泌儿茶酚胺的副神经节瘤患者常合并神经-皮肤综合征(neurocutaneous syndrome),如肌张力低下-毛细血管扩张症、结节性硬化、Sturge-Weber综合征等。Carney三联征(Carney triad)主要包括胃平滑肌肉瘤、肺软骨瘤和分泌儿茶酚胺的副神经节瘤,偶尔伴有食管平滑肌瘤和肾上腺皮质瘤(无功能或分泌皮质醇)。
单侧肾上腺嗜铬细胞瘤多为散发性而双侧肿瘤多为家族性
98%的嗜铬细胞瘤位于腹内和腹膜后,其中90%在肾上腺内。肿瘤的直径常小于10cm,多为3~5cm,平均重量10g左右,偶可超过1000g。肿瘤多为圆形或椭圆形,极少数为哑铃形;瘤体切面为灰色或棕褐色,或杂色相间,常有出血、坏死、囊性变或钙化。光镜下可见肿瘤由较大的多角形嗜铬细胞组成(下图);电镜下可见细胞核周围有密集的富含肾上腺素和去甲肾上腺素的嗜铬颗粒。恶性嗜铬细胞瘤的体积较大,可有包膜浸润或血管内瘤栓形成,但单凭显微镜所见很难鉴别,主要是观察其有无局部浸润和远处转移。转移的主要部位常为肝脏、骨骼、淋巴结和肺部。
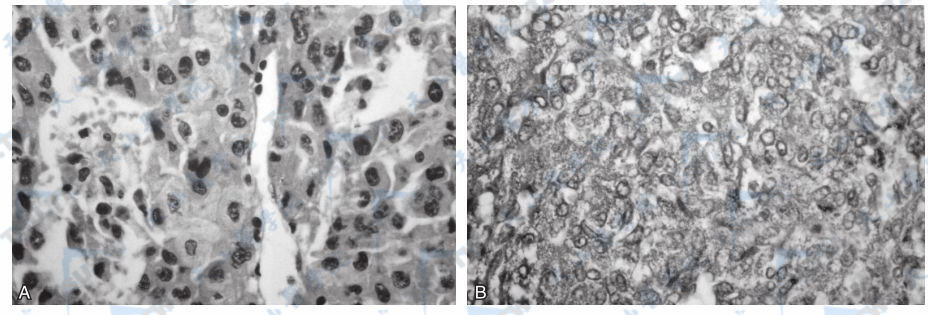
肾上腺嗜铬细胞瘤
注:A:瘤细胞大,胞浆丰富,核圆。瘤细胞被血窦分成巢状(HE法);B:切片铬粒素A(chromogranin A)染色,瘤细胞呈阳性反应(SP法)
家族性嗜铬细胞瘤常为双侧多结节性多中心病变,其恶性的发生率和复发率较散发型高。即使病理报告为良性嗜铬细胞瘤的患者术后也存在肿瘤转移可能(病理良性-临床恶性嗜铬细胞瘤);与此相反,病理学上表现为恶性特征,但其临床转归为良性过程(病理恶性-临床良性嗜铬细胞瘤)。因而,嗜铬细胞瘤术后的随访十分重要。
肾上腺髓质增生是嗜铬细胞瘤的特殊类型
肾上腺髓质增生主要指嗜铬细胞的数目增多,按肾上腺髓质/皮质厚度比值计,如>1∶10可认为有髓质增生(单纯性或伴MEN-2),大部分单纯性增生分布于双侧肾上腺髓质,少数为单侧性。一些免疫组化指标可用来判断肿瘤细胞的生物学行为。例如,单克隆抗体MIBI阳性细胞率在良恶性嗜铬细胞瘤中的差别很大,肾上腺的良性肿瘤细胞的MIBI阳性率低(0.81%)、恶性时增高(3.30%);在肾上腺外,这种差别更为明显(0.44% vs 5.1%),故当MIBI阳性细胞率>2%时,要高度疑为恶性嗜铬细胞瘤。
肾上腺外嗜铬细胞瘤分布广泛且生物学行为不一
家族性副神经节瘤的常见部位在头颈部,少数在胸、腹、盆腔或膀胱。奇怪的是,腹部的副神经节瘤分泌儿茶酚胺的可能性是头颈部副神经节瘤的10倍以上。多数肾上腺外嗜铬细胞瘤单发,部分为多发。约占全部病例的10%,但恶性可能性大。直径常小于5cm,重20~40g。肿瘤与肾上腺外嗜铬组织的解剖分布一致;大部分在腹部,可位于腹膜后腹主动脉前、左右腰椎旁间隙、肠系膜下动脉开口处或主动脉旁嗜铬体(Zuckerkandl器),亦可见于颈动脉体、颈静脉窦、肾上极、肾门、肝门、肝及下腔静脉之间、腹腔神经丛、胰头、髂窝、卵巢、膀胱、直肠后等处。胸部肿瘤常位于纵隔后交感神经干,也可位于心包或心脏;马尾及其他部位的肿瘤罕见。约20%肾上腺外嗜铬细胞瘤为多发性,恶性概率较大。用免疫组化方法可从瘤细胞中鉴定出肾上腺素、甲氧肾上腺素、去甲肾上腺素、甲氧去甲肾上腺素、多巴胺、血清素、乙酰胆碱、脑啡肽、CGRP、CRH、VIP、PACAP、ANP、AM、SS、神经肽Y、P物质和甘丙素等。一般来说,肾上腺嗜铬细胞瘤的多激素分泌特点较肾上腺外嗜铬细胞瘤明显;肿瘤细胞呈铬粒素、Leu7和S-100蛋白阳性反应仅说明其为神经外胚胎层来源,不能鉴别其良恶性。在细胞生长、浸润行为模棱两可,确诊有困难时,可借助流式细胞仪诊断。如仍困难,则需依赖于临床长期追踪观察。
本病的一般组织病理学诊断原则和方法可参照全美病理医师学院癌症委员会公布的诊断草案进行。
