
人类Y染色体是G组的近端着丝粒染色体,长约60Mb,仅大于21号和22号染色体。在不同种族、民族人群中Y染色体的长度具有很大的变异性。Y染色体是性染色体,为正常男性独有,由两个遗传功能不同的部分组成(下图)。拟常染色体区(pseudoautosomal region,PAR)和Y特异区(male specific region of the Y chromosome,MSY)。拟常染色体区位于Y染色体两端即PAR1和PAR2,在男性减数分裂过程中,参与同X染色体的配对、同源重组和分离。Y特异区占Y染色体全长的约95%,过去认为不参与同X染色体的同源重组,故又称为非重组区(non-recombining region,NRY),但也有研究表明该区同X染色体也有同源重组发生。
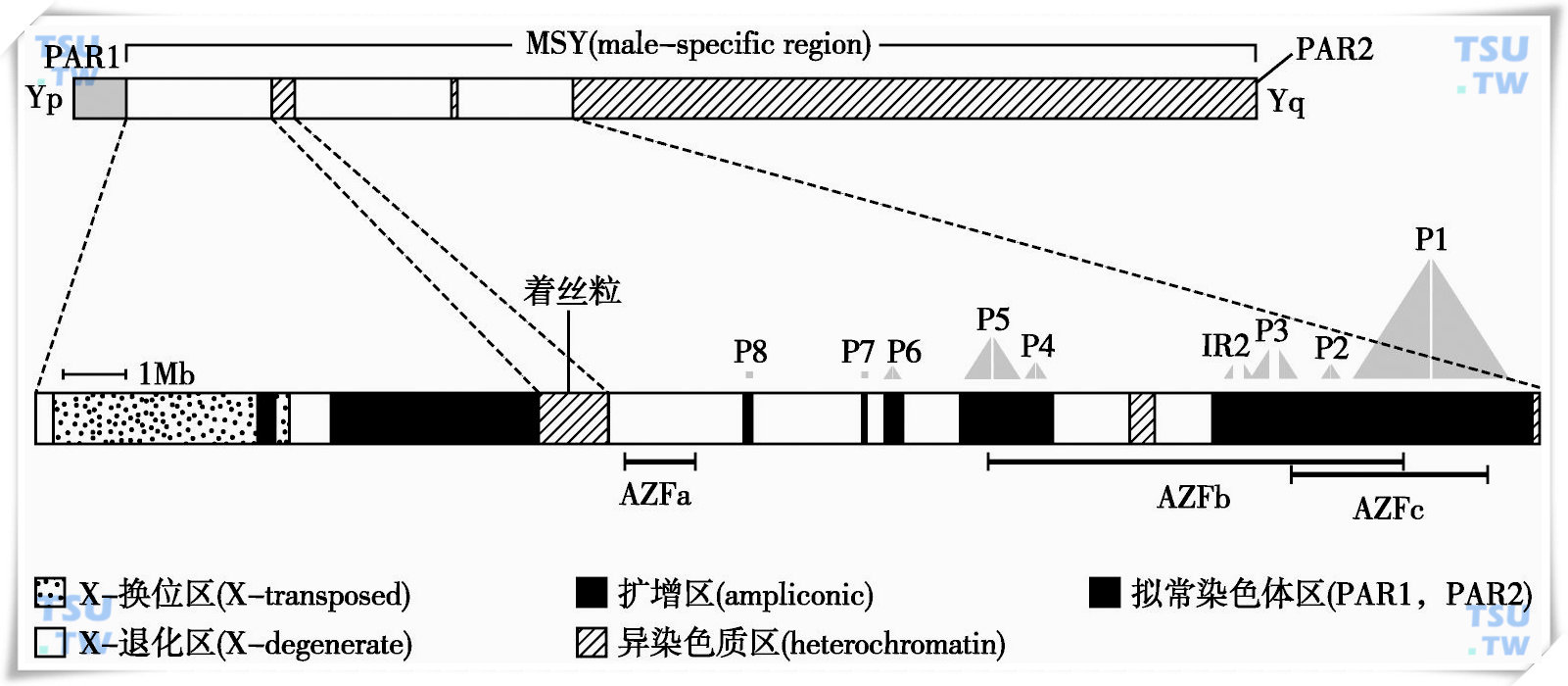
人类Y染色体结构图
Y特异区由异染色质区和常染色质区构成。异染色质区共有三段:长臂末断与PAR2临近段,长约40Mb;着丝粒,长约1Mb;新近Skaletshy等在对长臂常染色质区测序时,在中间发现了一段长约400kb的异染色质区,是由3000多个长达125bp的串联重复序列构成。Y特异区的常染色质区又根据系列的特点及其与X染色体的关系分为换位区(X-transposed)、X退化区(X-degenerate)和扩增区(ampliconic)。X换位区位于Y染色体短臂,与PAR1仅有一段短的X退化区相隔,并被一段扩增区隔成两段,总长约3.4Mb。该区的系列与Xq21有99%是相同的,被认为是3~4百万年前人类与猩猩分支时,由X染色体转位至Y染色体,故命名为X换位区。X退化区散在分布于Y染色体的短臂与长臂,共有27个与X染色体相对应的单拷贝基因和假基因,同源性60%~96%。可能是X、Y染色体共同从常染色体进化的遗迹。扩增区也呈散在分布,总长约10.2Mb,该区命名的根据是存在非常相近的系列,存在几十kb甚或几百kb系列99.9%相同。扩增区最大的系列结构特点是有8大回文结构系列,这些回文结构长9kb~1.4Mb,8大回文结构系列总长达5.7Mb,占Y特异区总长的约四分之一。扩增区还有反向重复顺序(inverted repeat,IR),主要有三个(IR1,IR2,IR3),重复顺序长度为62~298kb,有99.66%~99.95%相同。
无精子因子(azoospermia factor,AZF)基因定位于Y染色体的长臂区域6中,即Y11,控制精母细胞的发生,Chandley等认为AZF基因有助于胚胎发育过程中原始生殖细胞的分化。不同的男性群体可见不同的AZF基因型:如45,XO男性,46,XY正常男性和46,XX不育男性。Anderson等用Y特异DNA探针研究3例45,XO男性发现Y染色体的部分易位到了染色体14或15号上,每个易位的Y染色体节段均包括Yp、着丝粒区域和Yq的常染色质区段,尽管每个患者的断裂点均不相同。一个带有区域1~6的男性是可育的,他的SRY基因和AZF基因均存在;第二个患者具有区域1~5,有SRY基因,但没有AZF基因,该患者为无精子症不育男性;第三个患者是一个尿道下裂的男孩,他只有区域1~4B。综上所述,AZF定位在Y染色体的区域6,Y-常染色体的易位可以导致男性表型的发育,甚至在45,XO男性中导致精母细胞发生。
育龄夫妇中约有10%不能生育,其中一个重要的原因是男方生精障碍,表现为严重少精子(<2× 106/ml)或无精子,约占不育男性的10%。除输精管道梗阻、感染等病因外,遗传性状改变也是一个重要因素。Tiepolo和Zuffardi等于1976年发现6例无精子症患者均有Y染色体长臂的缺失,包括远端的荧光区和与其相连的非荧光区,因此推测在Yq11上存在着“Y染色体精子生成基因”。由于许多男性无精子症患者均有Yq11的异常,故将其称为AZF,以后许多学者的研究也证明了这一推测。1986年,Vergnaud应用Y染色体的质粒克隆DNA作为探针,分析Y染色体明显异常的患者,包括46,XY的男性,真两性畸形和Y染色体长臂缺失的病例,建立了Y染色体的基因缺失图谱;为Y染色体上的基因分析创造了条件。接着,其他一些学者也建立了Y染色体基因缺失图谱,并列出了Y染色体上各DNA片段的PCR引物序列,为用PCR方法检测Y染色体上的精子发生基因带来了极大的方便。他们发现,无精子症患者的基因缺失均发生于Y染色体的长臂,主要位于Yq11.23区域,而且并非是单一基因,可能是含有多个基因的大家族。目前已证明Yq11.23区域中多个基因片段的丢失均可引起生精障碍,但哪一个或哪几个基因是AZF尚不确定。
1996年,Vogt等在更大范围内对无精子症和严重少精子症患者进行了研究,他将Yq11区域再划分为25个段落,自Yq11的近侧端到远侧端分别命名为D1~D25,每个段落内选择1~7个不等,共76个DNA片段,用PCR的方法对370例无精子症和严重少精子症患者进行了检测,结果发现12例患者有基因片段的丢失,其中3例患者的丢失片段在Yq11的近侧端(D3~D6),3例患者的丢失片段在中段(D13~16),其余7例患者的丢失片段在Yq11的后段(D20~D22),因此认为在这三个区域内均存在AZF,分别称为AZFa、AZFb和AZFc。
AZF的重要候选成分
1992年我国留英学者马昆(Ma Kun)用特异探针在50例核型正常的无精子症或严重少精子症患者中,发现4例出现Yq11.23区域上有基因片段的微小丢失(microdeletion),认为这4人的无精子可能是由这些基因的丢失引起的。由于这4人丢失的DNA片段并不重复,相互没有重叠,证明AZF区域很大或不止一个基因,甚至是位于Yq11.23区域上的一个基因家族。接着用相对患者缺失的一段DNA序列作为探针,自Y染色体质粒库中筛选获得一系列质粒克隆,其中用Y367探针筛选获得A5F克隆,再用A5F DNA探针筛选人睾丸cDNA文库,筛出2株阳性克隆,命名为MK5和MK29,均为1.9kb左右,其中MK5为1878bp,推算编码蛋白含496个氨基酸。MK29为1874bp,与MK5相比,少了4个核苷酸,并另有7个核苷酸不同,推算编码蛋白含419个氨基酸,由于7个核苷酸不同,使得肽链中的3个氨基酸也不同,在两个DNA序列中均含有多聚A结尾,查阅基因库未查到相同序列,用MK5探针Northern Blot检测睾丸组织中mRNA,发现一条识别带,约2kb,而在脑、心、肾、肝、脾、前列腺、肌肉等组织中均未见识别带,Southern Blot识别不同物种的基因组DNA,仅在人可见识别带,证明MK5高度专一地在人睾丸内表达。应用基因库(gene bank)进行基因结构分析,发现MK5和MK29均有编译含RNA识别位点蛋白质的序列,因此将MK5和MK29分别命名为YRRM(Y chromosome RNA recognition motif)1和2,提示在Y精子发生初期,该基因可能参与RNA的形成、加工和翻译等过程。此外,在YRRM的基因产物中,含有以Ser-Arg-Gly-Tyr氨基酸为单位的多次重复序列,由于在RNA的翻译启动因子序列及其他一些RNA结合蛋白序列中均有这个重复序列,再次证明YRRM基因对精子发生过程有重要的调节功能。用原位杂交方法定位YRRM1于Yq11.23区域上,睾丸切片原位杂交还显示YRRM1基因主要在精原细胞和初级精母细胞阶段表达。用Southern blot杂交技术检测无精子症患者的基因组DNA,发现2例有YRRM1基因的丢失,进一步用PCR方法扩增YRRM基因,也发现了2例无精子症患者有YRRM的丢失。日本学者选用马昆的YRRM引物,PCR检测日本男性基因组DNA,发现日本人中无YRRM2基因,可能系人种差异,在正常男性中,均检出YRRM1基因,在63例核型正常的无精子症男性中,3例有YRRM1缺失,间接证明了YRRM对精子发生作用,但其调节机制至今尚不明确。江苏省生殖医学重点实验室应用YRRM1引物PCR方法检测了130例核型正常的无精子症或严重少精子症患者基因组DNA,发现10例患者有YRRM1的丢失,丢失率为7.7%。但Reijo的研究显示,在89例患者中未见YRRM基因的丢失,因此他认为YRRM可能不属于AZF的候选成分。
除YRRM外,DAZ基因是AZF的又一重要候选成分。1995年,Reijo等人在Y染色体上随机选择了84个DNA片段,用PCR的方法对90例有生育能力的男性和89例原因不明的无精子症和严重少精子症患者进行了检测,90例正常男性均无基因片段的丢失,89例患者中,12例有基因片段的丢失,丢失片段的大小不一,但均位于Yq11的远端,检测这12例患者的父亲和兄弟的基因组DNA,均未见丢失片段,说明基因片段的丢失可能是造成这些患者精子生成障碍的原因,对这些患者进行睾丸活检,未发现生精障碍的组织学变化与基因片段丢失。为获得具有表达功能的AZF候选成分,Reijo用AZF区域内的不同探针筛选人睾丸cDNA文库,获得了一个阳性克隆,称为DAZ(deleted in Azoospermia),并证明原有基因缺失的12位患者均有DAZ的丢失,而他们的父亲和兄弟均未见DAZ的丢失。Southern blot证明DAZ为人和猩猩所特有,但在人的电泳图上显示二条杂交带,其中一条为男女都有,以后证明为DAZ的同源基因,位于常染色体上。Northern blot证明DAZ专一地在睾丸内表达,DAZcDNA的基因结构分析显示其编码蛋白含366个氨基酸,蛋白质的相对分子质量约41 257,氨基端的氨基酸顺序类似于RNA结合蛋白的氨基酸顺序,提示DAZ的表达蛋白可与RNA或单链DNA结合,从而调节RNA或DNA的功能。目前已发现果蝇和小鼠体内也有DAZ的同源基因,分别命名为Boul基因和Dazh基因,已证明Boul基因是果蝇的精子发生基因,Boul基因转移入有生精基因缺陷的果蝇可以明显促进生精功能。Dazla基因位于小鼠的17号染色体上,仅在小鼠睾丸内特异表达,且表达时间和强度与精子发生的时间相一致,用转基因的方法将变异的Dazla基因导入小鼠体内,对携带变异基因的纯合子小鼠,精子不能发生,对携带杂合子的小鼠,则精子生成减少,直接证明了Dazla基因对精子发生的作用,为深入研究DAZ基因的特性和功能意义创造了条件。此外,在人的常染色体上也有一个同源基因,称为DAZH,其编码蛋白含295个氨基酸,相对分子质量为33 170,氨基酸排列顺序与DAZ的编码蛋白相似,但不完全相同,原位杂交显示DAZH位于3号染色体短臂的末端,Northern blot显示DAZH也特异地在人睾丸内表达,但DAZH与精子发生的关系尚不明确。
Kobayashi等研究也发现,除YRRM和DAZ外,在Y染色体上还含有多个与生精相关的基因片段,发现在50例正常核型的无精子症患者中,有5例有基因片段的丢失,丢失片段均位于Yq11.23区域内的DYS7C和DYS1之间,另一例患者只有DYS7C的丢失,进一步在该区域内选择15个基因片段,分别为DYS7E、DYS230、DYS224、DYS226、DYS7C、DYS233、DYS232、DYS1、DYS236、DYS237、DYS238、DYS240、DYS239、DYS247、DYZ1,用PCR进行扩增。在63例核型正常的无精子症患者中,有9例至少有一段基因的丢失,其中有8例丢失片段位于DYS7C~DYS239之间,另一例丢失片段较大,DYS226~DYS134,该患者同时有YRRM1的丢失,提示YRRM1位于DYS134和DYS226之间,结果表明在DYS226~DYS247至少有一个基因是生精必须的,特别是DYS1与DAZ极为相似,可能DYS1就是DAZ。
AZF缺失的发生率
各实验室报道的AZF在无精子症和/或严重少精子症患者中的发病率差异较大,从3%~29%,多数集中在13%~15%,造成这种差异的原因有多种,包括:①病例选择,对于严格选择病例仅对不明原因的无精子症或严重少精子症患者进行检测,则缺失率较高;②实验方法包括实验条件的好坏,影响到结果的可靠性;③检测不同的候选成分也会得出不同的缺失率,如仅检测DAZ基因,则缺失率较高;④人种差异,如西方男性有YRRM2基因,日本人则没有。
AZF缺失与睾丸病理的关系
对这些患者进行睾丸活检,发现睾丸的病理改变与AZF丢失部位有明显的相关性,其中AZFa丢失的患者最为严重,表现为唯支持细胞综合征(Sertoli-cell-only syndorme),同时有睾丸体积的缩小,AZFb丢失的患者表现为生精阻滞,主要停留在精母细胞阶段,睾丸内可见精原细胞和初级精母细胞,但没有精子生成,以上两类患者的精液检查,AZFc丢失的患者情况比较复杂,对于同一患者,一些生精小管内仅见支持细胞,另一些生精小管内可见精原细胞、精母细胞、精子细胞甚至精子,精液检查表现为严重少精子,有时可见少量活动精子,但畸形精子明显增多。
AZF与严重少精子症的关系
AZF的丢失不仅可以导致无精子症,也可引起严重的少精子症,马昆已发现一例少精子症患者有YRRM1的丢失,1995年,Iwamoto等亦报道1例严重少精子症患者有AZF的丢失,丢失区域为DYS7C~DYS239,Reijo等1996年报道,在35例严重少精子症患者中有2例发生AZF的缺失,且丢失片段较大,而且与多数无精子症患者的基因缺失部位相同。Vogt研究发现,AZF丢失发生在Yq11远侧端时常导致少精子症。本室用PCR方法检测了25例严重少精子症患者的YRRM1基因,亦发现了1例患者有YRRM1基因的丢失,因此,对于严重少精子症患者,也有进行AZF检测的必要。
AZF缺失与ICSI
对于严重少精子症或者精液中无精子但睾丸内有单倍体的生精细胞患者,可用单精子卵细胞胞质内注射(intracytoplasmic sperm injection,ICSI)的方法使其获得生育能力,但这种助孕方法有可能将精子带有的基因缺陷传给子代。已证明只要能受精,AZF的缺失并不影响子代的生长发育,但男性子代仍是AZF缺陷患者,成年后也将表现为无精子或严重少精子症,因此对于这些患者是否应该用ICSI的方法助其生育仍是有争议的一大问题。无论如何,在行ICSI治疗或植入前进行AZF缺陷的诊断都是有意义的,也是必须的。
