
英文:short-rid-polydactyly syndrome;
MIM:263530、263520、2635610;
同义名:短肋-多指侏儒征、Saldino-Noonan综合征,多指伴新生儿软骨营养不良型。
溯源:1971年首由Majewski报道,1972年Saldino和Noonan又陆续报道。1977年Nauwott等亦有报道,并于1978年将其分为三型。其特征是肋骨发育不全或不发育,并有多指(趾)畸形。
发病机制
病因不明。基本的遗传缺陷尚未确定。骨切片可见骨发育不良十分显著,软骨被过早的骨化所破坏,生长排列着少见的肥大性软骨细胞,而且增生与退化的交织使生长带几乎不能辨认,在某些地方生长带实质上缺如。干骺端含有成骨细胞、破骨细胞和过早的骨化、空泡等,十分混杂。电镜下并未见软骨细胞和间质细胞形态学的变化,软骨细胞培养表明与正常软骨细胞超微结构无明显差异。
遗传学:各家报道的家族病例多为同胞患病,故认为遗传方式呈AR遗传。唯Richardson曾报道Ⅱ型的家系病例,认为该型属AD遗传,目前遗传方式尚未定论。
临床表现
特征分型
本病分Ⅰ、Ⅱ、Ⅲ型。骨骼畸形是本征的主要特征:肋骨短缩呈水平状,胸廓窄小以至使充满胸腔的肺重量只有9g,四肢短小甚至呈鳍状四肢,X线甚至见管状骨呈卵圆形。干骺端发育不良,早期骨化、多指(趾)、赘生并指(趾)多发于尺(腓)侧。除上述程度不同的共有临床表现外,三种类型所伴有的其他系统异常为:
- Ⅰ型(Saldino-Noonan综合征),骨骼畸形较另两型严重,鳍状肢体显著;颅顶、脊椎、骨盆等亦有骨化不全;管状骨干骺端由于发育不全而非常不规则;小额骨、髋臼扁平常有骨刺突向中间和侧面,常没有唇、腭裂或会厌畸形(图a~c)。
- Ⅱ型(Majewski综合征),最具特点的表现是短得极不相称的胫骨和管状骨干骺端边缘较规则,以及多发性并指畸形,骨盆和脊柱却正常,唇、腭裂、会厌畸形,内脏畸形有多囊肾、大血管转位、肺发育不全、胃肠道和泌尿系闭锁或畸形,两性生殖器或性别分辨不清、小脑蚓部缩小,脑回粗大沟深。有的还有肝大和腹水(图d-f)。
- Ⅲ型(Nauwott综合征),除Ⅱ型所具有的内脏畸形外,颅骨和脊椎畸形更为常见,如短颅底,前额膨大,鼻梁下陷,枕骨扁平等。另外颜面水肿,呼吸道异常,垂体功能低下等亦有报道。
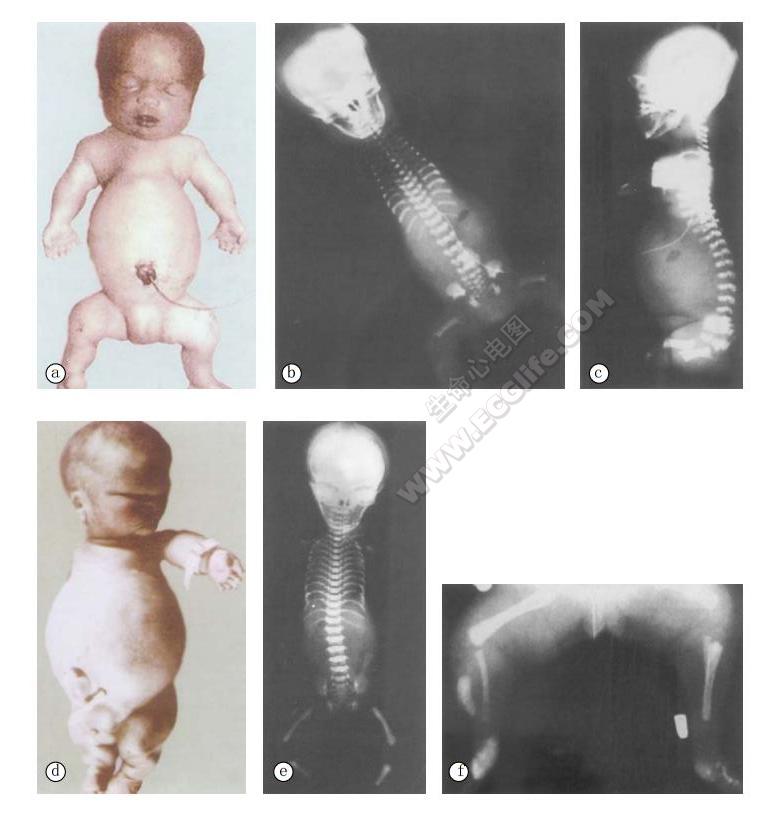
- a:女性死婴,为Ⅰ型(Saldino-Noonan型)患者。身材矮小,水肿样外观。颅骨、脊椎、骨盆骨骨化不良;肋间距短,胸廓窄,多指(趾);
- b、c:X线显示胸廓狭窄,肋骨短,肋骨呈平行走向,长骨干骺端不齐,中段、末端有骨刺;
- d:新生女婴,为Ⅱ型(Majewski型)患者;短肢型身材矮小,中线唇裂、腭裂,蒜头鼻(短鼻),耳小、畸形,胸廓狭窄,锁骨高位,肋骨短,轴前多指,短指(趾);
- e、f:X线显示胸廓狭窄,肋骨短、平等。胫骨不对称缩短。
心血管损害
最为常见,外显率高达50%以上,以Richardson等报道的9例中有8例伴有心血管畸形。主要以大血管转位和肺动脉狭窄最常见。此外室间隔缺损,主动脉狭窄,右心发育不良及右室双出口等均可见到。
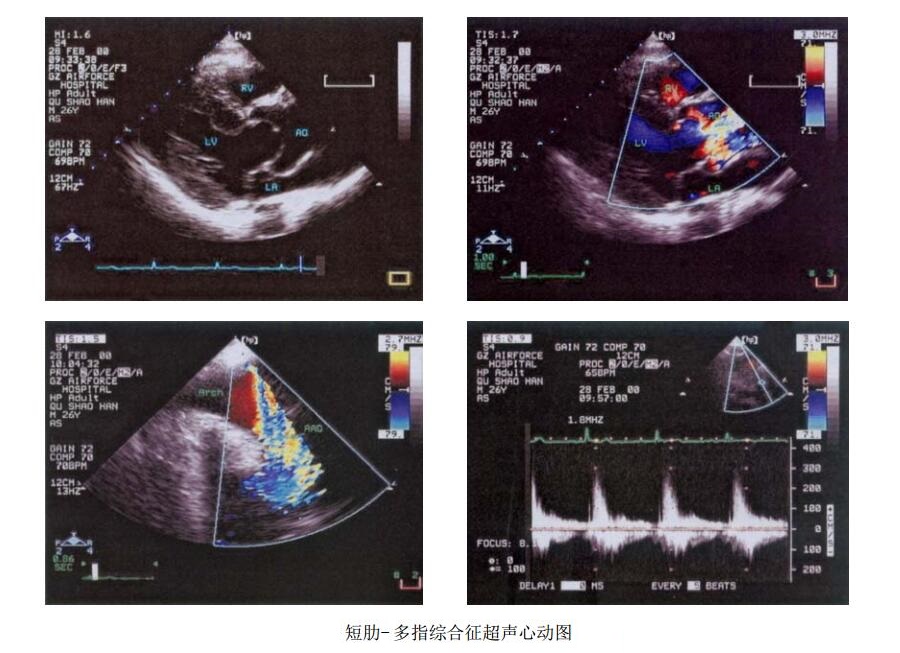
- 左室长轴断面:因瓣口开放受限,呈圆顶状凸向升主动脉,升主动脉内径增宽。
- CDFI:示起源于狭窄主动脉瓣口的蓝色为主五彩明亮的射流束;主动脉弓长轴断面:显示升主动脉腔收缩期高速花色射流;连续血流频谱:主动脉瓣上峰值速度为3m/s。
- 超声提示:先天性主动脉瓣狭窄。
诊断、治疗及预后
- 诊断:依据新生儿发病,短肋、短肢、多指(趾)并内脏畸形,诊断即可成立。
- 治疗及预后:近年来采用X线、胎儿镜和超声波检查如在妊娠期20~40周前仍不能发现胎儿长骨之存在则可做出产前诊断,应终止妊娠。目前尚无有效疗法,本征预后不良,患儿多在出生前或出生后1周内死亡,自发生流产和死胎的发生率颇高。
系统的医学参考与学习网站:天山医学院, 引用注明出处:https://www.tsu.tw/heart/zhz/xiantian/qita/755.html
