
英文:Huichinson-Gilford syndrome
同义名:早老症( progeria)、小儿早老症、早老矮小病、早衰综合征、Gilford垂体性幼稚型侏儒征、淋巴体质性侏儒征、Gilford综合征、Vriot-Pironneau综合征。
一种较罕见的常染色体隐性遗传性疾病,1886年首次由Hutchinson报道,1897年Cilford尸检1例,并详加描述而得名。此后,屡有报道。其特征为出生时正常,其后生长逐渐迟缓,呈侏儒伴各种早老表现。
发病机制
早老症的病因尚未完全阐明,可能与遗传因素或炎症、损伤等引起脑垂体前叶功能不足有关,也可能因为结缔组织细胞不能对生长激素起反应,以致胶原发生老化。2003年美国的科研人员发现一种叫Lamin A的蛋白突变可以造成这种早衰症,最近的研究表明Lamin A突变会使DNA受损修复系统丧失功能,从而导致早衰现象的出现。另有研究提示,端粒长度变短和端粒酶活性异常可能在HGPS早老的致病过程中起重要作用。细胞遗传学分析表明,同卵双生的HGPS患者皮肤活检提示70%的细胞存在染色体异常46XY, inv ins(1;1)(q32;q44q23),提示与HGPS有关的基因可能位于插入染色体片段的断裂处。
临床表现
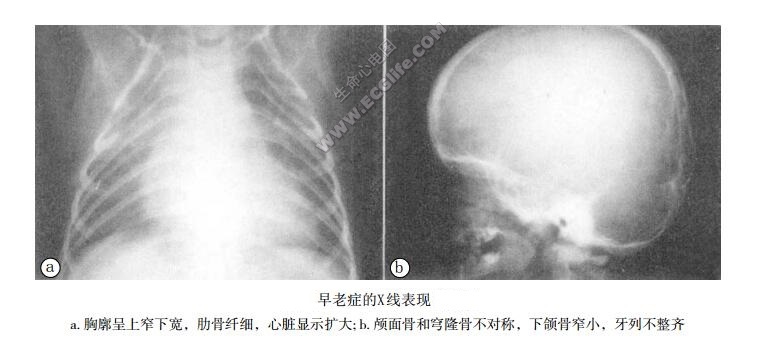
本病多在2岁左右发病,多在青春期死于心脏供血不足。临床上病儿呈侏儒状,明显消瘦,前额突出,面部狭小,小下颌,呈典型的鸟样面容。皮下脂肪消失,肌肉萎缩,牙齿排列不齐,牙龈萎缩。患儿的坐行走均晚于同龄正常儿。患儿表现进行性衰老,早期心血管系统可以正常,以后出现进行性动脉硬化,可累及冠状动脉和脑动脉。其X线表现主要为全身骨质疏松,锁骨短小,肋骨和长骨纤细,颅面骨和穹隆骨不对称,下颌骨窄小,牙列不整齐,腰椎可呈鱼口状表现。
心血管损害:主要为动脉硬化,可累及冠状动脉与脑动脉。
诊断、鉴别
患者于幼年即出现生长发育迟缓,具有早老的特殊性体型与面貌,且合并高血压、心脏退行性病变等疾患,一般不难诊断。
本征应注意与垂体性侏儒、Werner综合征及Turner综合征鉴别。垂体性侏儒身材矮小,但无早老面容及早年心血管病。Werner综合征又称成人早衰症,为常染色体隐性遗传疾病,一般在青春期后发病,全秃不常见下颌骨完全正常,多合并白内障、硬皮病等疾患。Turner综合征与性染色体异常(45,XO)有关,表现为卵巢与外生殖器不发育,原发性闭经。
治疗、预后
治疗无特殊方法,主要用维生素及降脂药物,控制动脉硬化的发展。2006年美国《Science》杂志发表的一篇文章中,Loren Fong等用一种药物阻止早老症小鼠模型体内对lamin A蛋白的修饰,从而减轻了这些小鼠的骨质疏松等类似早老症的症状,而且在这个短期的研究中,这些小鼠的寿命延长了一些。目前这一药物正在作为抗肿瘤药物在进行临床试验,这为早老症患儿的药物治疗带来了一线希望。
早老症预后不良,常在20岁之前因并发症死亡,存活年龄的中位数为12岁。
