
英文:Hurler syndrome (OMIM 252800);
同义名:I-H型、黏多糖贮积症I-H、多发性骨发育不良综合征(dysostosis multiplex syndrome)、Pfaundler-Hurler综合征。
溯源与发展
1917年首由Hurler报道一家系兄弟2人患病,又于1919年就两个患儿具有某些相同临床表现,因以病因不明,根据丑陋面貌采用承留病等加以命名。其后Pfaundler等详加报告因而得名Hurler综合征、Hurler-Pfaundler综合征。本征是黏多糖贮积症中的常见类型。
- 遗传学:系谱分析呈AR遗传方式。致病基因定位于4p16.3。
新生儿中发病率为1/10万,杂合子频率为1/150。患者多数在10岁前死亡。
发病机制
原发缺陷再于溶酶体内缺乏α-L-艾杜糖苷酸酶(α-L-lduronidase)缺乏。此酶缺乏使酸性黏多糖硫酸皮肤素(dermatan sulfate,即硫酸软骨素B)及硫酸乙酰肝素的降解受阻而贮积,在组织细胞内堆积形成Hurler细胞,为组织病变的基础。尿中可检出硫酸皮肤素和硫酸乙酰肝素。
神经系统、肝、网状内皮系统、内分泌腺、软骨等结缔组织基质中有大量黏多糖堆积。心脏受损很常见且很严重,临床表现很突出。
临床表现
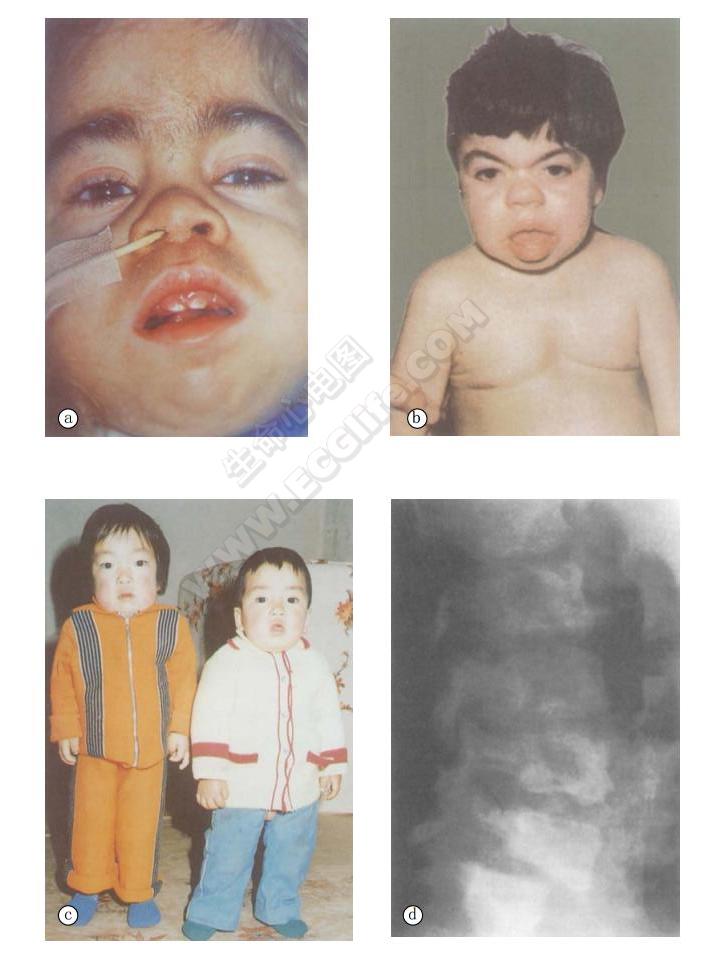
呈侏儒痴呆状、头大、貌丑,角膜混浊(corneal clouding),视力障碍,眉毛浓而且成连眉或称一字眉(synophrys),眼距增宽,鼻梁平塌,鼻孔宽而前倾,张口,唇舌大,牙小而疏,皮肤粗糙,尤其是上肢及胸部,可呈结节样增厚,毳皮多,满布全身。骨骼异常,胸部畸形,胸腰部驼背,四肢短,掌宽而手指粗短,手指部分屈曲,似爪形手,关节僵硬,活动受限。渐进性智力发育不全和体质损害,进行性肝脾肿大。
- 心血管损害:二尖瓣受损,近乎100%,其次为三尖瓣、主动脉瓣、肺动脉瓣。瓣膜增厚,边缘粗糙,腱索增厚并缩短。50%~80%患者有冠状动脉硬化、管腔狭窄。较大的周围动脉管腔亦有类似改变。心外膜、心包亦可受损,而心内膜、心肌损害少见。
临床表现为心脏增大,各瓣呈关闭不全,可有心绞痛,高血压及心律失常,最后发生充血性心力衰竭。
辅助检查
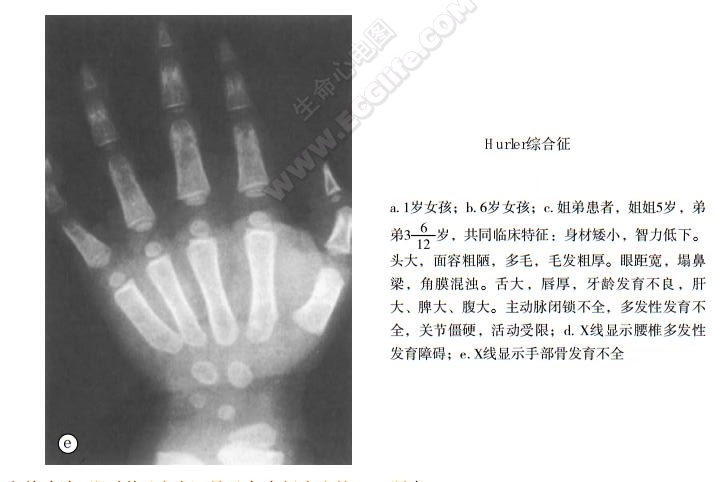
- X线示心脏扩大,肺动脉高压、肺淤血表现。颅骨大,额骨及枕骨骨质肥大、蝶鞍增大而浅;脊柱后突,长骨髓腔扩大等改变。
- 超声心动图示心肌肥厚、二尖瓣关闭不全等改变。
- 心电图无特征性改变,可见右室肥厚等。
- 化验尿中可检出大量硫酸皮肤素及硫酸乙酰肝素;血中淋巴细胞和粒细胞有Reilly小体。
诊断
根据本征临床特征性外貌表现,100%心脏瓣膜改变及进行性心力衰竭;X线改变,尿检黏多糖增多,即可诊断。
治疗及预后
无特殊方法。宜防止感染、加强保护及对症治疗。维生素A醇( Retinot)疗法可使成纤维细胞内黏多糖减少,其疗效有待进一步证实,骨髓移植疗效尚无定论,心脏手术效果不佳。
产前确诊,即时终止妊娠,是避免本征出生的最好的措施。
本征预后极差,约50%病例死于心力衰竭,10岁前多因反复心肺并发症导致功能衰竭而死亡。
