
毛细胞白血病(HCL)是一种慢性恶性淋巴细胞增殖性疾病,以外周血和骨髓中出现毛细胞、脾大而无浅表淋巴结肿大为特征。本病由Bouroncle等于1958年首先以白血病性网状内皮细胞增生症报道。此后,又以慢性网状淋巴细胞增多症、脾髓质组织淋巴细胞增生症、网状细胞增多症、恶性网状细胞增多症、非白血病性网状细胞增多症等相继报道。毛细胞白血病这一名词由Schrek和Donnelly于1966年提出。
病因及发病情况
本病的病因尚不清楚。欧美人发病率明显高于亚洲人。美国洛杉矶地区1972~1987年间年龄调整后的发病率为男性2. 9/106,女性0. 6/106,男女之比为4. 8:1;英国约克郡地区1985~1990年20~79岁人群的年发病率为2. 9/106(男性发病率为4. 0/ 106,女性为1. 7/106,男女之比为2. 35:1),45岁以上者发病率明显增高。
临床表现
本病以中老年人多见,中位发病年龄在50岁以上。最常见的症状为乏力、左上腹部不适。常见体征为脾脏肿大,发生率为80%~90%,肝脏肿大不如脾脏明显,浅表淋巴结肿大少见。贫血、出血、感染、体重下降、纳差也是较常见的病征。约1/4的患者伴有自身免疫性疾病。较罕见的病征尚有骨损害,多发生于股骨近端,病损类似于多发性骨髓瘤,系毛细胞浸润所致。此外,脾破裂、皮肤及中枢神经系统浸润亦有报道。下表列举了意大利HCL协作组725例HCL的临床表现。
意大利HCL协作组725 例HCL的临床特点
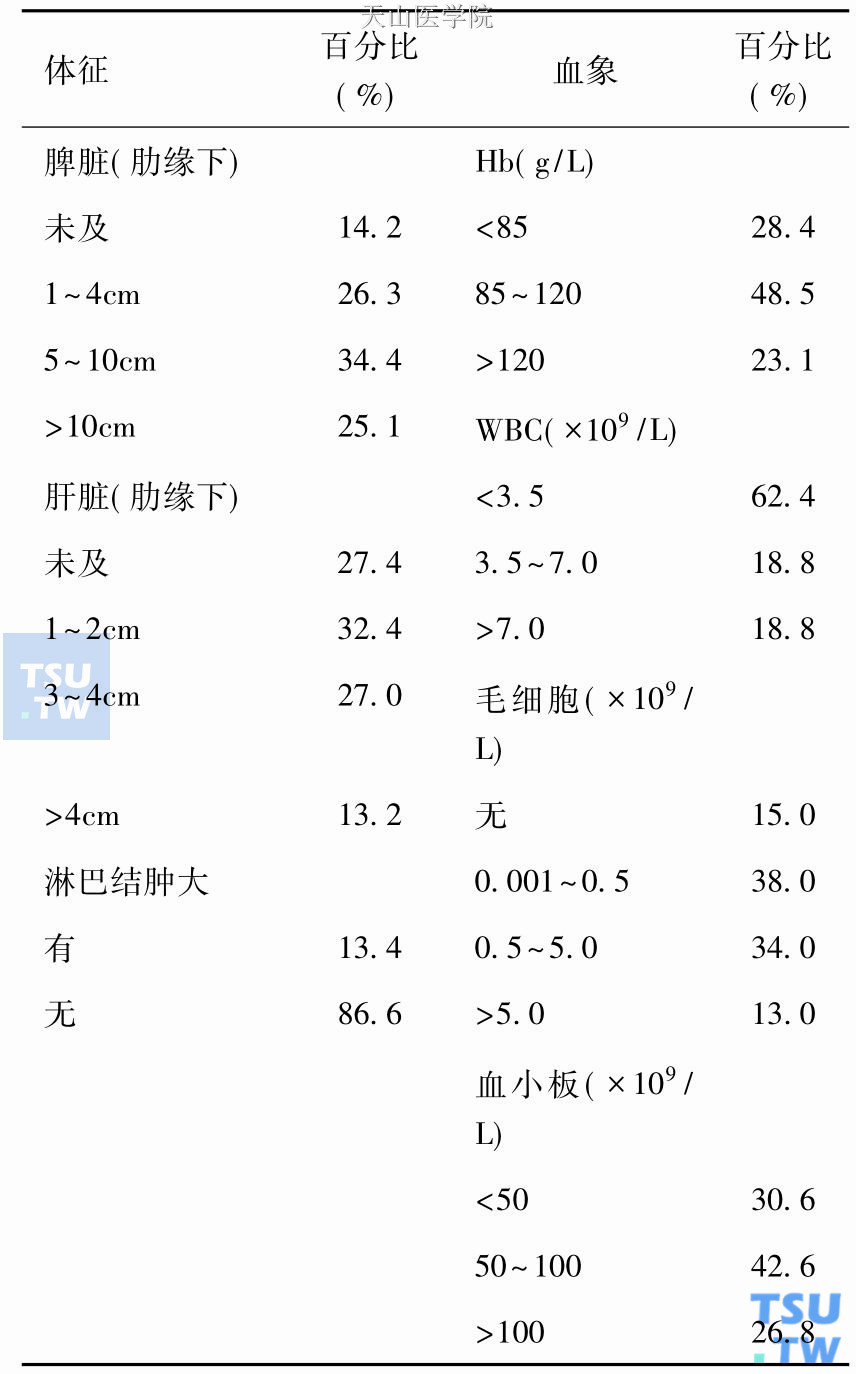
引自Frassoldati A等,Leukemia and Lymphoma,1994,13:307
分型与临床分期
Jansen根据血红蛋白(Hb)水平和脾脏大小对本病进行分期,标准如下:
分期
Ⅰ期:Hb>120g/L,脾脏肋缘下≤10cm;或Hb>85g/L,脾脏肋缘下<4cm。
Ⅱ期:Hb>120g/L,脾脏肋缘下>10cm;或Hb 85~120g/L,脾脏肋缘下4~10cm;或Hb<85g/L,脾脏肋缘下<4cm。
Ⅲ期:Hb 85~120g/L,脾脏肋缘下>10cm;或Hb <85g/L,脾脏肋缘下>4cm。
脾切除3个月以上者:
A期:Hb>120g/L,中性粒细胞(NC)>0. 5× 109/L。
B期:Hb>120g/L,NC≤0. 5×109/L;或Hb 85~120g/L,NC>0. 5×109/L。
C期:Hb 85~120g/L,NC≤0. 5×109/L;或Hb<85g/L。
分型
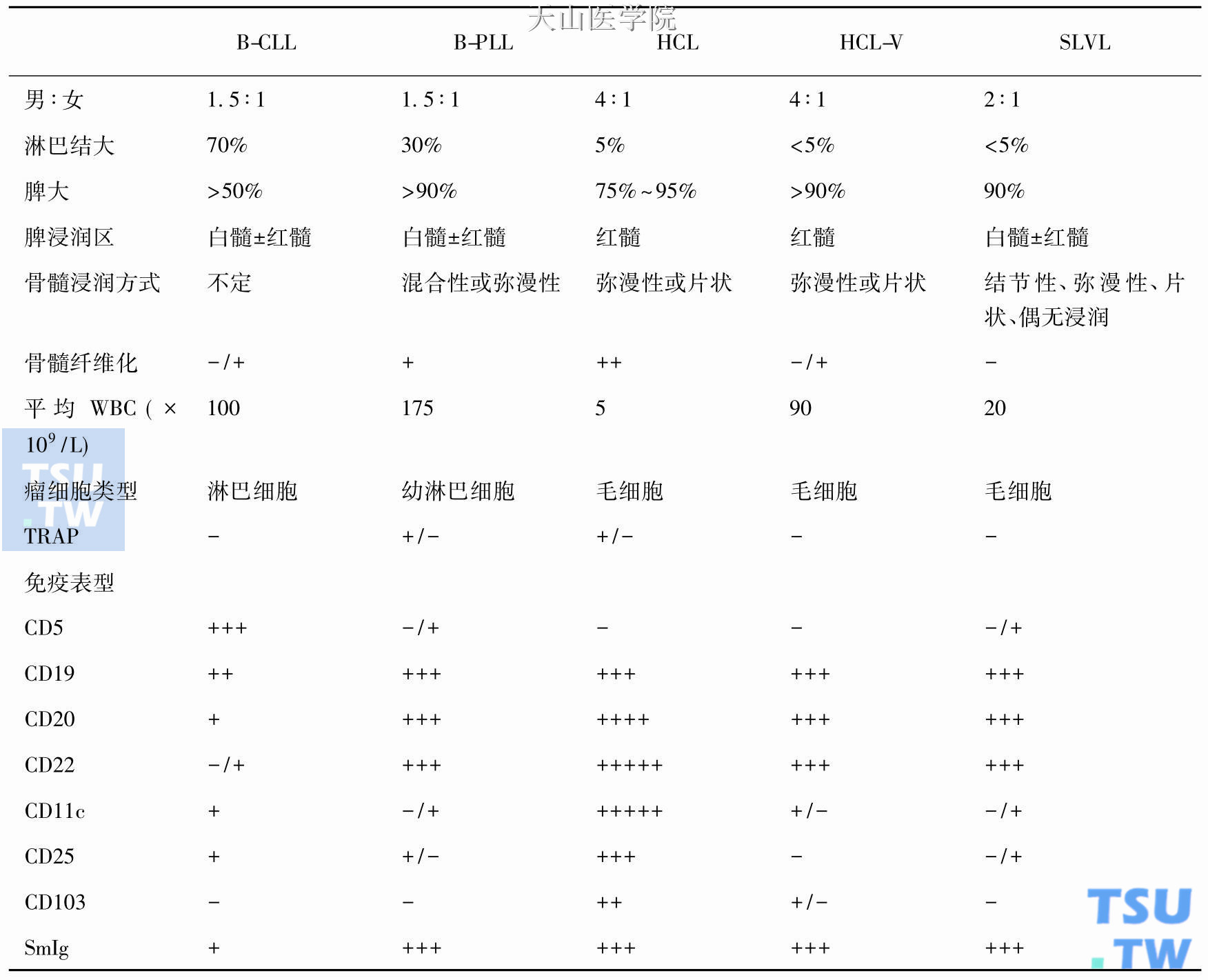
本病分为典型HCL和变异型HCL两种类型。各自特点见下表。另外,东方人HCL与西方人HCL比较有一些不同的临床特点:东方人HCL全血细胞减少较少见,白细胞增高较常见,骨髓干抽较少见,耐酒石酸酸性磷酸酶(TRAP)多为弱阳性或阴性,核糖体板层复合物(RLC)少见。
HCL及HCL- V与B- CLL、B- PLL及SLVL的鉴别
实验室检查
所有患者骨髓都有毛细胞浸润,外周血毛细胞检出率可达90%。单核细胞减少、血小板减少、贫血常见,半数以上有中性粒细胞减少,近半数可有全血细胞减少。细胞遗传学检查提示,1q42、2q11和5q13异常可能是本病较特征性的改变。
病理学特点
骨髓:低倍镜下毛细胞均匀分布,胞核与胞核分隔开,核周有丰富而浅淡的胞质围绕,整体呈现一种疏松的“星空样”或“蜂窝样”改变,高倍镜下毛细胞胞质丰富,胞质突起清晰可见,胞质浅淡,细胞核周围呈透亮区,核卵圆形或有凹陷,核染色质疏松,核仁不明显,核分裂象极少见。增生活跃的骨髓可有残留造血灶,多为小簇状红系或散在的巨核系造血灶,粒系造血灶少见。网状纤维常增多。此外,可见大量红细胞外渗形成血窦。变异型HCL与典型HCL的主要区别在于前者毛细胞胞质量较少,胞质突起纤细,分布不均匀,核圆形,折叠或双核,染色质致密,核仁明显。
脾脏:以红髓浸润为主,白髓多萎缩或消失,可有假窦形成。毛细胞形态与骨髓一致。
肝脏:大多数患者均有肝脏受累,主要累及肝窦和门脉区。
淋巴结:尸检时大多数患者有纵隔、腹腔或腹膜后淋巴结肿大,皮质区、副皮质区和髓质区均有毛细胞浸润。
此外,肺脏、肾脏、胃肠道、中枢神经系统、皮肤等均可发生毛细胞浸润,但非常少见,一般在患者死后尸检时才发现。
超微结构:毛细胞胞质突起有两类:①微绒毛状,为纤细的指状突起,直径50~150nm,长约0. 6~1. 4μm;②伪足状,基底较宽的舌状突起,宽约1μm,长约3μm,细胞质内高尔基体发达,线粒体丰富,粗面内质网丰富,可见溶酶体颗粒,约半数可有RLC。
细胞化学:最显著的特点是酸性磷酸酶(ACP)强阳性,且TRAP阳性。但亚洲国家HCL患者的TRAP多为弱阳性或阴性。因此,TRAP阴性不能排除HCL。
咐醇酯(TPA)诱导分化试验:毛细胞在体外与TPA孵育后迅速贴壁,镜下见胞质伸出长枝状突起,其他白血病细胞无此特点。
细胞免疫表型见上表。
诊断与鉴别诊断
本病迄今尚无统一的诊断标准,诊断主要依靠:①临床特点;②外周血和骨髓中发现毛细胞;③毛细胞的细胞化学、超微结构和免疫表型;④病理组织学特征。
本病主要应与CLL、PLL、SLVL、单核样B细胞淋巴瘤(MBCL)、边缘区B细胞淋巴瘤(MZBL)等相鉴别:SLVL、MBCL、MZBL和HCL的瘤细胞对应的正常细胞相同或相似,前三者的瘤细胞均为胞质丰富的单个核细胞,胞质突起较少,染色质致密,常有一个明显的核仁。在组织切片中,这些细胞与毛细胞非常相似,但染色质致密、胞质突起短而少有别于典型毛细胞。三者的免疫表型与HCL相似,但TRAP常为阴性或弱阳性。此外,组织学特点与HCL不同:MZBL骨髓浸润方式为片状或局灶性,无网状纤维增生,脾脏浸润以红髓为主;MBCL骨髓受累少见且多见于小梁周围,外周血受累罕见,淋巴结浸润多见于滤泡间区或窦间隙。与CLL、PLL、SLVL的鉴别见上表。
此外,本病还应与恶性组织细胞病,全身性肥大细胞病等相鉴别。
治疗
治疗指征:①巨脾导致压迫症状;②Hb<100g/L;③PLT<100×109/L;④WBC>20×109/L;⑤中性粒细胞绝对值小于1. 0×109/L伴反复机会性感染;⑥出现自身免疫并发症;⑦组织毛细胞浸润。
若无上述指征者,不需治疗,但需定期随访,以监测病情进展。
治疗方案:
一、干扰素α(IFN-α):为HCL的首选药物。用法:2×106U/m2,3次/周,共12个月。有效率为80%,CR率9%。主要副作用为流感样症状。
二、核苷类似物:有喷司他丁(DCF)、克拉屈滨(2- CdA)和氟达拉滨。
DCF用法:4mg/m2,静脉输注,隔周一次,共3~6个月。主要副作用为感染及免疫抑制。
2- CdA用法:0. 1mg/(kg•d)×1周,静脉输注。主要副作用为发热及免疫抑制,但比DCF轻得多,有可能取代IFN-α成为HCL的首选药物。
氟达拉滨的用法为30mg/(m2•d)×5天,每个月1疗程。对HCL也有效,但远不及2- CdA。这三种药物的疗效见下表。
核苷类似物对HCL的疗效
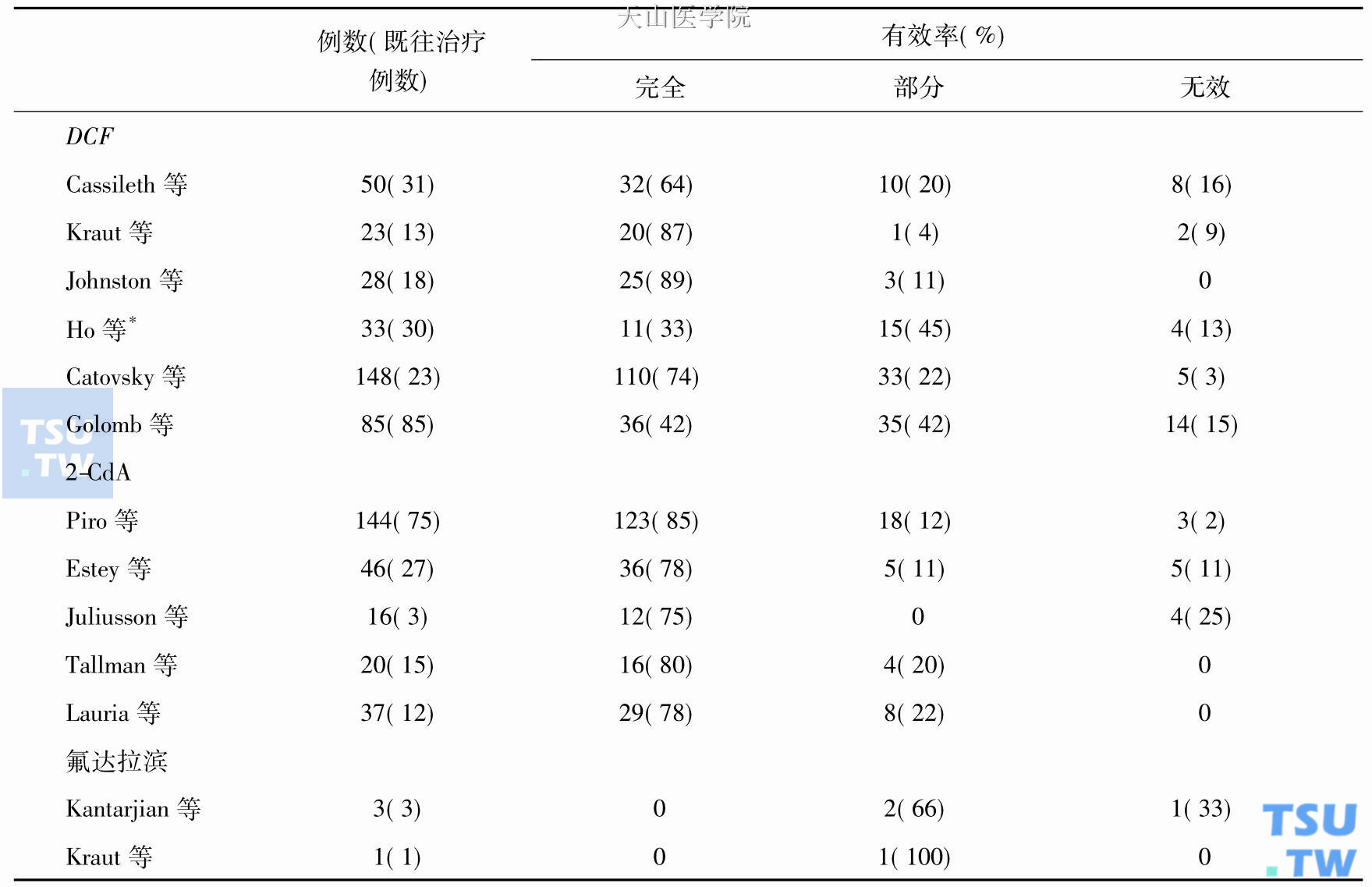
*其中3例因早期死亡而舍去
引自Tallman MS等,Blood,1995,86:2463
三、脾切除:在干扰素及核苷类似物问世之前,脾切除是本病的首选治疗手段,现已不主张对HCL患者施行脾切除术,但对2- CdA,DCF和IFN-α无效或巨脾产生压迫症状或脾破裂者可考虑。
四、其他:在干扰素问世前,苯丁酸氮芥及联合化疗均曾用于脾切除后病情恶化的患者,但疗效均不理想,目前已不作为一线治疗方案。由于药源及费用等原因,我国目前仍较多选用苯丁酸氮芥作为一线药物。
疗效标准:
CR:外周血及骨髓中毛细胞消失;无脾脏肿大,血象恢复正常:Hb>120g/L,PLT>100×109/L,中性粒细胞>1. 5×109/L。
PR:骨髓中毛细胞数下降50%以上,血象恢复正常至少1个月。
微效:外周血象指标至少一项恢复正常。
进展:Hb<100g/L,无出血征象,中性粒细胞计数连续3次低于1. 5×109/L或PLT<100×109/L。
预后
自从IFN-α,DCF和2- CdA用于治疗HCL后,HCL的预后明显改善,其5年存活率达87. 5%。感染是HCL的主要死因,严重粒细胞缺乏者常发生败血症或肺炎。由于单核细胞减少,NK和T细胞功能缺乏,导致细胞免疫缺陷,因而HCL患者发生机会性感染的危险也增加。
