
肿瘤恶液质患者肌肉快速降解的机制仍不清楚。有认为肿瘤细胞或者宿主细胞释放某些代谢介质参与了恶液质的过程。骨骼肌细胞内ATP-泛素化蛋白质降解途径的激活是由宿主和肿瘤衍生的相关介导因子引起。
蛋白质降解诱导因子
蛋白质降解诱导因子(proteolysis-inducing factor,PIF)。PIF是一种分子量为24kD的硫酸化糖蛋白。从MAC16肿瘤分离的PIF或胰腺癌恶液质患者尿液分离的PIF都可迅速降低小鼠体重,24小时内体重下降达10%,并且专一性消耗瘦肉组织,小鼠腓肠肌下降64%,比目鱼肌下降17%,而心和肾没有影响,肝脏重量增加。PIF对骨骼肌影响主要同时通过抑制蛋白质合成和增加蛋白质降解。研究发现PIF特异性增加腓肠肌泛素、E214k和C9蛋白酶体亚基的表达,即通过增加泛素-蛋白酶体通路分子表达促进骨骼肌蛋白的降解。在离体鼠肌小管研究中确定了PIF激活蛋白质降解和抑制蛋白质合成导致肌肉萎缩的信号通路见图3-4-8。PIF可诱导小鼠肌小管泛素-蛋白酶体通路组分表达,包括20S蛋白酶体α亚基、MSS1、P42、19S调节因子ATPase亚基,以及增加蛋白酶体β5亚基的类糜蛋白酶活性。研究揭示PIF作用需要通过激活转录因子NF-κB信号通路。IκB激酶β(IKKβ)/ NF-κB 对于诱导泛素-蛋白酶体通路激活发挥重要作用,通过小鼠肌肉专一性表达活性IKKβ激活NF-κB可引起类似临床恶液质的肌肉消耗,包括E3连接酶MuRF-1表达升高3.3倍,蛋白酶体C2和C9亚基表达升高2.4~2.8倍;激活NF-κB还可抑制肌源性转录因子MyoD表达,导致肌球蛋白合成下降。同时用白藜芦醇治疗可明显减轻荷瘤小鼠恶液质状况,包括能明显缓解体重下降和肌肉蛋白降解等,研究证实白藜芦醇通过抑制IKK而抑制NF-κB激活。这进一步验证了NF-κB激活对于恶液质肌肉萎缩的重要意义。PIF除了增加蛋白酶体组分表达和激活外,还同时增加PPⅡ表达。PIF激活NF-κB信号过程中还涉及活性氧簇(ROS)产生,这可能是许多引起肌萎缩的共同步骤(图3-4-8)。
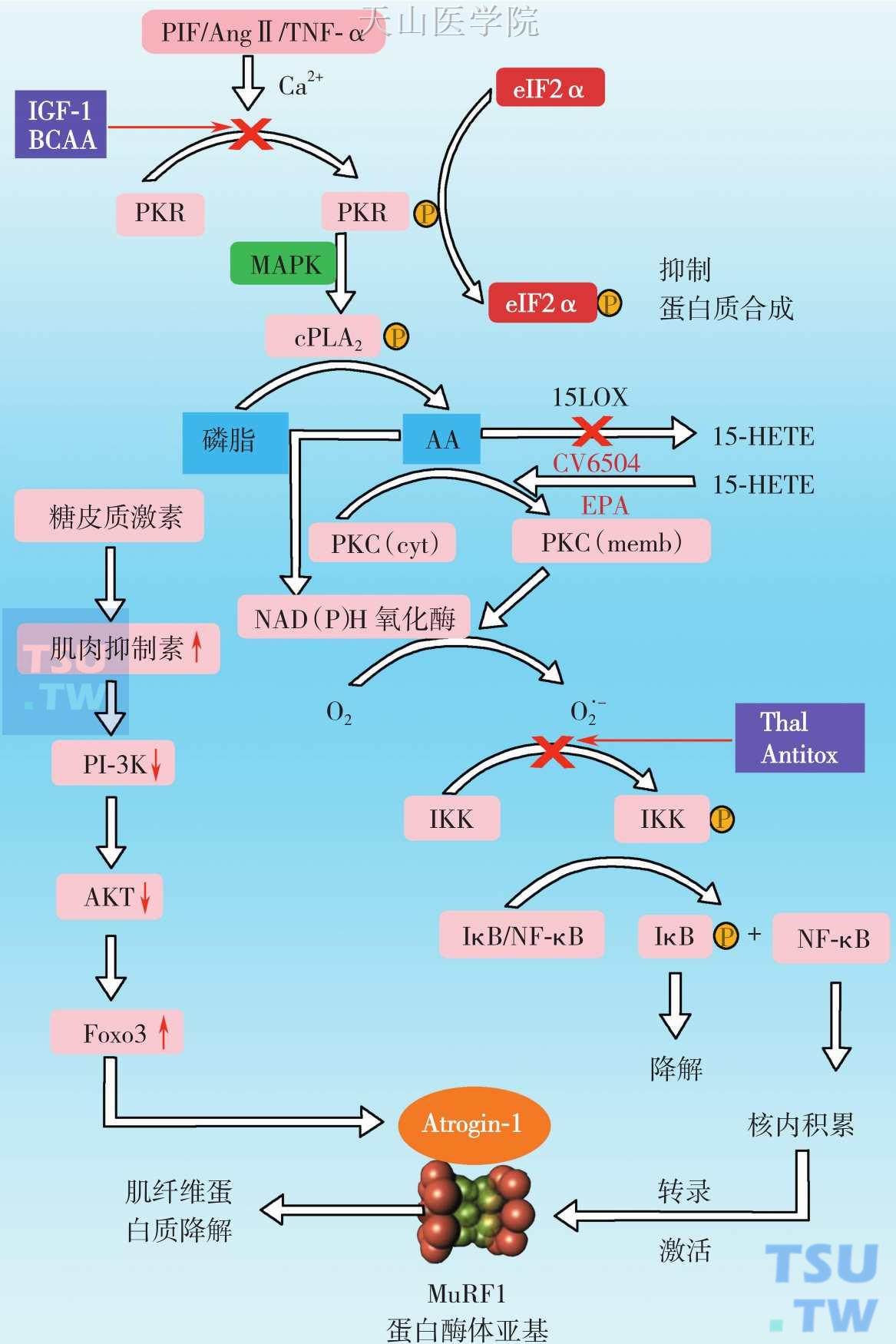
图3-4-8 恶液质肌肉高代谢和萎缩的机制
缺乏抗氧化酶Cu/Zn-SOD的小鼠呈现明显加快年龄相关的肌肉萎缩,过氧化氢等氧化应激可通过诱导泛素-蛋白酶体组成表达而导致小鼠肌小管蛋白降解。PIF通路产生ROS还涉及PKR磷脂酶A2(PLA2)、花生四烯酸(AA)、NADPH氧化酶和蛋白激酶C(PKC)(图3-4-8):PTF诱导产生ROS需要激活NADPH氧化酶,该酶活性需要AA激活,而AA则由PLA2水解膜磷脂2位酯键而释放并进入胞质,胞质中的AA不仅可激活NADPH氧化酶,还可激活PKC,以及直接作用线粒体产生ROS。PKC激活对于PIF激活NADPH氧化酶也是必需的,PKC还可能涉及15-脂肪氧合酶(15-lipoxygenase,15LOX)转化AA生成15-羟二十碳四烯酸(15-hydroxyeicosatetrienoic acid,15-HETE),因为15LOX抑制剂可明显减轻鼠恶液质模型的肌萎缩程度。PKR是PIF通路上游蛋白激酶,PKR一方面通过磷酸化修饰激活PLA2,而激活蛋白降解通路,同时还可以磷酸化修饰eIF的α亚基而抑制蛋白翻译。因此,PKR是连接骨骼肌蛋白降解和蛋白质翻译抑制的重要分子,PKR抑制剂能通过增加蛋白质合成和抑制蛋白质降解机制明显减轻恶液质鼠模型骨骼肌萎缩,同时发现PKR抑制剂还能抑制肿瘤生长。BCAA和胰岛素样生长因子1 (IGF-1)增加磷酸酶PP1表达使PKR脱磷酸而失活,从而发挥减轻恶液质蛋白降解作用。PIF受体是一个分子量为40kD膜蛋白,目前已经明确了骨骼肌和肝脏细胞表达,而脂肪组织和肾没有发现。研究证明PIF受体的抗体(针对肽链N-末端抗体)在体外明显阻断PIF的效应,并且在鼠恶液质模型上显著减轻骨骼肌萎缩。除了作用于骨骼肌外,PIF还可直接诱导肝脏产生细胞因子,使用重组人PIF可以诱导人肝脏内皮细胞和脐带静脉内皮细胞释放IL-6和IL-8,还可诱导人巨噬细胞和单核细胞释放TNF-α、 IL-6和 IL-8,这些效应是通过NF-κB 和STAT3 转录通路实现的。PIF还可以诱导共结合蛋白聚糖从人静脉内皮细胞脱落,该效应与肿瘤转移和恶化密切相关。
细胞因子
细胞因子如TNF-α、IL-6、IL-1 和IFN-γ等在肿瘤恶液质过程中扮演重要角色。许多来自动物研究证实TNF-α在恶液质肌肉萎缩中发挥重要作用。Oliff A等将转染人TNF-α基因的中国仓鼠卵细胞进行小鼠移植后产生类似恶液质症状,表现为进行性消瘦、厌食和过早死亡等;Llovera M等将Lewis肺癌细胞种植进Ⅰ型TNF-α受体缺陷小鼠体内后,小鼠骨骼肌消耗明显改善,显著好于野生对照组动物,野生鼠骨骼肌消耗伴有骨骼肌蛋白降解率的明显升高,而实验鼠没有明显改变,同时两组动物体内蛋白质合成没有发生变化。而当用重组的TNF-α处理动物可增强腓肠肌蛋白质分解和降低蛋白质合成。TNF-α主要通过类似PIF方式增加ROS导致肌肉蛋白降解,已证实TNF-α可引起骨骼肌氧应激和一氧化氮合酶增高,用抗氧化剂和一氧化氮合酶(nitric oxide synthase,NOS)抑制剂处理可以防止动物体重下降、骨骼肌消耗和骨骼肌分子异常等。同样,TNF-α也是通过NF-κB而激活泛素-蛋白酶体通路,TNF-α还可以通过ROS激活p38MAPK系统,而后者被认为是骨骼肌分解代谢的潜在调节因子,对于骨骼肌专一性基因表达是必须的。此外,TNF-α也可以通过AA和LOX代谢物来诱导ROS产生(图3-4-8)。TNF-α还可通过NF-κB诱导MyoD降解而抑制肌肉生成。
转基因过表达IL-6的小鼠也出现骨骼肌萎缩,同时伴有组织蛋白酶B和L,以及泛素(多聚体和单体)表达增高,有IL-6受体抗体干预可以阻断上述效应。一种多发性结肠癌小鼠模型(ApcMin/+)中,血浆中IL-6水平最高的小鼠整体恶液质症状和多发性肠癌最明显,而ApcMin+/IL-6-/-小鼠肌肉没有萎缩和肠癌较小。这提示IL-6通过增加肿瘤负荷来诱发恶液质的。体外研究肌小管发现IL-6同时激活泛素-蛋白酶体通路(增加26S蛋白酶体)和溶酶体途径(组织蛋白酶B和L)来促进长寿命蛋白质降解和降低其半衰期。
其他研究发现肿瘤坏死因子相关弱凋亡诱导因子(TWEAK) 是一个潜在的骨骼肌消耗的细胞因子,研究结果提示 TWEAK可通过抑制PI3K/Akt信号通路、激活NF-κB系统以及泛素-蛋白酶体系统引起骨骼肌萎缩。
血管紧张素Ⅱ
临床研究表明血管紧张素Ⅱ(angiotensinⅡ,AngⅡ)可以促进骨骼肌蛋白分解,用血管紧张素转化酶(angiotensin converting enzyme,ACE)治疗充血性心衰时可以增加恶液质患者皮下脂肪和肌肉,而注入ANGⅡ可引起小鼠体重明显下降,并以瘦肉组织丢失为主。体外研究表明ANGⅡ是通过增加泛素-蛋白酶体通路表达和活性来促进肌肉蛋白降解的。体外研究还显示ANG Ⅱ可抑制肌小管蛋白质合成,同时注入ANGⅡ小鼠血浆和肌肉组织内IGF-1明显降低。而过表达IGF-1可以对抗ANGⅡ引起的骨骼肌萎缩,以及抑制泛素连接酶Atrogin-1 和MuRF-1表达。因此,与PIF类似机制 ANGⅡ通过 PKR激活泛素-蛋白酶体通路抑制蛋白质合成和促进蛋白质降解,其他还包括通过PKC→NADPH氧化酶→ROS→NF-κB (图3-4-8)。而IGF-1对抗ANGⅡ作用机制主要诱导PP1表达,使PKR脱磷酸而抑制NF-κB和泛素-蛋白酶体通路激活,同时也通过降低eIF-2的α亚基磷酸化而减轻蛋白质合成的抑制作用。体外研究提示IGF-1对抗ANGⅡ作用还通过Akt/mTOR/p70S6k的信号通路。
糖皮质激素
糖皮质激素由于其有效缓解恶液质症状(改善食欲、增加食物摄入和减轻不适感觉等)成为恶液质的辅助用药,因为其引起骨骼肌萎缩(主要影响Ⅱ型肌纤维)的副作用,所以只能限制在癌症终末期应用,而且只能短期应用。尽管切除肾上腺并不能改善动物恶液质,但糖皮质激素在恶液质发展过程中可能发挥重要作用。糖皮质激素通过上调泛素-蛋白酶体通路引起肌肉萎缩,该作用是通过叉头型转录因子(Foxo)而非上述因子激活的NF-κB。专一性骨骼肌过表达Foxo1转基因小鼠体重和骨骼肌质量明显下降,并且缺失Ⅰ型和Ⅱ型肌纤维,增加Atrogin-1表达,而MuRF-1不变,同时伴有溶酶体组织蛋白酶L上调。相反下调Foxo1可以增加恶液质小鼠肌肉质量,提高MyoD和降低肌肉抑制素水平。小鼠肌小管中激活Foxo3可通过溶酶体途径和泛素-蛋白酶体途径促进蛋白质降解,并且溶酶体途径为主。糖皮质激素激活Foxo是通过降低IGF-1/PI3K/Akt而减少Foxo磷酸化来实现的(图3-4-8)。
糖皮质激素还可激活肌肉其他转录因子,如CCAAT/增强子结合蛋白(C/EBP)-β和-δ,以及激活因子蛋白-1(AP-1)。C/EBP可以结合在鼠泛素、C3蛋白酶体亚基和E214k基因的启动子区。这些转录因子在恶液质骨骼肌中是上调的,并在肌肉萎缩中发挥重要作用。钙也参与糖皮质激素诱导的肌肉蛋白降解作用。因为用钙螯合剂BAPTA或钙调蛋白激酶Ⅱ抑制因子KN-62能显著改善由地塞米松引起的肌肉蛋白降解。钙还在调节钙调蛋白酶活性中发挥重要作用,研究提示增加钙蛋白酶活性是肌丝拆解的一个早期的限速步骤,涉及Z-带结合蛋白即粗丝结合蛋白(Titin)和α-辅肌动蛋白降解,以及肌动蛋白和肌球蛋白释放。
糖皮质激素诱导肌肉萎缩还伴有肌肉内肌肉抑制素表达增加,而肌肉抑制素缺失可以阻止糖皮质激素效应。这提示肌肉抑制素在糖皮质激素引起肌萎缩中可能发挥重要作用,肌肉抑制素是TGF-β超家族成员,是肌肉生长的负调因子。过表达肌肉抑制素动物可以产生类似恶液质状态下的肌肉和脂肪严重丢失。有研究发现谷氨酰胺可以抑制肌肉抑制素表达,从而改善由糖皮质激素引起的肌萎缩。体外研究发现肌肉抑制素可抑制肌肉生成相关基因MyoD和Pax3表达,而促进泛素相关基因Atrogin-1、MuRF-1 和E214k表达,还可抑制Akt磷酸化从而增加Foxo1活性。这表明糖皮质激素引起肌萎缩的机制不同于PIF(图3-4-8)。
骨骼肌细胞凋亡参与恶液质肌肉萎缩
除了上述蛋白酶体和溶酶体途径外,肌肉细胞凋亡在肌肉萎缩中也发挥重要作用。Ishiko等提出肿瘤发展过程中宿主肌肉消耗的两个机制:肌肉细胞凋亡发生在早期而后期主要是代谢异常即肌肉的高代谢状态。许多动物模型结果证实肿瘤恶液质早期骨骼肌细胞凋亡活性增强的:荷瘤引起恶液质小鼠腓肠肌内凋亡蛋白酶(caspase)-1、-3、-6、-8和-9活性升高的,并出现多聚(ADP-核糖)聚合酶(PARP)片段化。在Yoshida AH-130腹腔积液肝肿瘤大鼠、Lewis肺癌小鼠和UX2肿瘤兔骨骼肌中发现凋亡典型特征的梯状DNA片段,促凋亡蛋白Bad在动物体重下降早期表达显著升高。近年来上消化道肿瘤伴有明显体重下降患者的骨骼肌尸检结果显示骨骼肌DNA片段显著升高,是对照组的3倍,同时伴有PARP片段化和MyoD蛋白水平下降。
总之,肿瘤恶液质发生机制十分复杂,涉及肿瘤类型、发展阶段和患者状态等,肿瘤本身是恶液质患者骨骼肌不断萎缩发生和发展的主因,其主要机制涉及肿瘤和宿主来源的细胞因子包括PIF、TNF-α、IL-6、IL-1和IFN-γ,以及ANGⅡ和糖皮质激素等,这些分子可以激活相关信号通路,包括PKR→PLA2→ PKC→NADPH氧化酶→ROS→NF-κB;肌肉抑制素→PI3K→Akt→Foxo等,最后激活骨骼肌蛋白降解通路,如溶酶体通路和泛素-蛋白酶体通路,同时抑制蛋白质合成,以及还可能激活骨骼肌细胞凋亡通路等。
