
家族遗传性垂体瘤与基因突变相关
约5%的垂体瘤为家族性,其中一半的家族性垂体瘤属于1型多发性内分泌腺肿瘤(multiple endocrine neoplasm type 1,MEN-1)和Carney复合症(Carney complex)的一部分;其他类型的家族性垂体瘤统称为家族性单一性垂体瘤(familial isolated pituitary adenomas,FIPA)。McCune-Albright综合征、多发性内分泌腺肿瘤综合征、Carney复合症、家族性GH瘤和家族性PRL瘤的病因已经基本明确,分别与Gsα、menin、1型α亚基蛋白激酶A(PRKAR1A)、AIP和p27(CDKN1B)基因突变有关。GH瘤和Carney复合症的发病与GNAS1种系突变的关系密切,而散发性垂体瘤编码的PRKAR1A活性增强。所以,似乎所有类型的垂体瘤均与环一磷酸腺苷信号(cyclic adenosine monophosphate pathway)增强有关。
FIPA的共同特点是:①发病年龄相对较小;②肿瘤体积较大;③约15%的患者存在芳(香)烃受体相互作用蛋白基因(aryl hydrocarbon receptor interacting protein gene,AIP)突变。
功能性垂体瘤与激素分泌失常相关
下丘脑功能紊乱
下丘脑促垂体激素和垂体旁分泌或自分泌激素可能在垂体瘤形成的促进阶段起一定作用。GHRH有促进GH分泌和GH细胞有丝分裂作用,分泌GHRH的异位肿瘤或转GHRH基因动物可引起垂体GH瘤。某些生长因子,如PTHrP、PDGF、TGFα、TGFβ、IL、IGF-1等在垂体瘤中有高水平表达,它们可能以旁分泌或自分泌方式促进垂体瘤细胞的生长和分化。神经生长因子(NGF)缺乏对于PRL瘤的发生和发展起一定促进作用。在正常腺垂体的发育阶段,NGF具有促进PRL细胞分化和增殖作用。
下丘脑抑制因子的作用减弱可促进某些垂体瘤的发生。Cushing综合征患者在作肾上腺切除术后,皮质醇对下丘脑CRH分泌的负反馈抑制减弱,CRH分泌增多,继而发生ACTH瘤(Nelson综合征),说明缺乏正常的靶腺激素负反馈机制及随后的下丘脑调节功能紊乱对垂体瘤的发生起了促发作用。
促激素分泌失常
PRL瘤、GH瘤、LH/FSH瘤、TSH瘤、ACTH瘤等的发病可能与相应的促激素(PIF、多巴胺、GHRH、GnRH、TRH、CRH等)分泌失常有关,由于促激素分泌过多或下丘脑释放因子/下丘脑释放抑制因子分泌失衡而导致相应垂体细胞增生。在特定条件下,相应垂体激素失去有效反馈抑制作用,也可形成结节或肿瘤。例如,异位肿瘤(ectopic tumor)分泌GHRH(支气管类癌、胰岛细胞瘤、小细胞型肺癌等,异位GHRH综合征)可引起垂体GH细胞增生或形成GH瘤,而异位肿瘤分泌CRH出现垂体ACTH细胞和GH细胞增生或ACTH瘤(异位CRH/ACTH综合征)。有时,非内分泌组织的恶性肿瘤还分泌MSH、LPH、CLIP、β-内啡肽,如果肿瘤分泌异位激素的功能够强,时间较久,则足以使垂体的相应细胞形成增生性结节或肿瘤。
靶激素分泌失常
靶激素分泌减少或靶激素对垂体的抑制作用减弱是功能性垂体瘤的重要原因。一般认为,由于相应的靶激素失去对垂体激素的有效反馈抑制作用而形成结节或肿瘤。例如,原发性甲减时,因T3/T4缺乏或对垂体TSH细胞的抑制作用有抵抗,TSH细胞呈代偿性增生肥大,并在一定条件下形成TSH的自主性分泌,严重者形成结节或TSH瘤。类似的情况亦见于肾上腺皮质功能减退症,特别是双侧肾上腺皮质切除后(Nelson综合征)。
多种假说解释无功能性垂体瘤的发病机制
无功能性垂体瘤(non-functioning pituitary adenoma,NFPA)的病因和发病机制未明,曾提出过多种发病机制假说。NFPA通常来源于LH/FSH细胞或ACTH细胞,虽然无激素分泌增多的临床表现,但其本质是LH/FSH瘤、ACTH瘤或GH瘤。因此,应将功能性垂体瘤和NFPA作为一个激素分泌谱来看待,它们的区别只是NFPA处于谱的最低处,而功能性垂体瘤处于该谱的最顶端。
NFPA的生物学特征
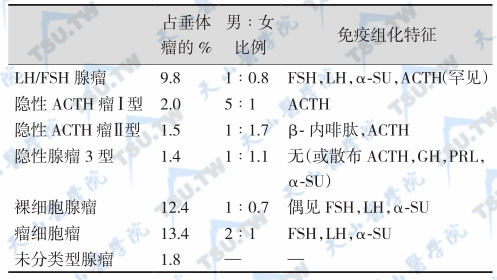
注:α-SU:glycoprotein α-subunit,糖蛋白α-亚基
无功能腺瘤的生物学行为
如表3-12-9所示,用免疫组化方法发现,无功能腺瘤的生物学行为有以下6种类型:
- 分泌无生物活性的糖蛋白激素α亚基(α-亚基瘤);
- 分泌生物活性的糖蛋白激素β亚基(β-亚基瘤);
- ACTH瘤,但因激素产生或翻译后的修饰过程存在缺陷,血液循环中ACTH仍正常(静止型ACTH瘤,silent ACTHoma);来源于ACTH细胞的NFPA表现为Golgi体的ACTH分泌颗粒包装障碍、细胞内分泌颗粒分泌无能、分泌的ACTH分子无活性(前体大分子)或ACTH翻译与翻译后缺陷(缺乏激素原转换酶)。
- LH/GH/ACTH阳性反应(隐匿型LH瘤/GH瘤/ ACTH瘤);
- 免疫组化阴性的裸细胞瘤(immunonegative null cell adenomas)。
- 腺垂体意外瘤(accidentenoma)无临床表现,仅在头颅影像检查时被意外发现。
无功能性垂体腺瘤还有两个较突出的生物学行为特点:一是虽然不出现激素分泌过多的临床症状,但在肿瘤体积达到一定程度时,因压迫垂体或脑组织而出现相应症状(20%~25%),其中最常见的表现是视觉损害和腺垂体功能减退;二是无功能腺瘤尽管存在生长抑素受体,但一般对生长抑素的反应很差,而对替莫唑胺(temozolomide)有较好疗效,其原因在于该药可影响DNA修复酶——0-6-甲基鸟嘌呤-DNA甲基转换酶(0-6-methylguanine DNA methyltransferase)的活性。
肿瘤抑制基因沉没和凋亡基因不适当甲基化
一般认为,垂体瘤与细胞获得增殖能力(尤其是肿瘤抑制基因失活)有关,一些研究提示,垂体瘤与甲基化(methylation)相关的肿瘤抑制基因沉没(gene silencing)或与垂体瘤凋亡基因(pituitary tumour apoptosis gene,PTAG)的不适当甲基化有关。肿瘤抑制基因沉没和凋亡基因不适当甲基化的病因未明,可能与下列因素有关:
- 遗传性因素:如MEN-1突变、垂体瘤转录因子prop-1过量、Carney复合症等;
- 下丘脑因素:如GHRH过量、CRH过多、某些下丘脑激素受体活化性突变等;
- 垂体因素:如某些信号转导分子(gsp、CREB)突变,或FGF-2、EGF、NGF等生长因子过多,癌基因激活、GHRH、TRH等;
- 环境因素:如E2、放疗;
- 靶腺(甲状腺、性腺、肾上腺)功能衰竭。
细胞缺陷和下丘脑调控失常
有关垂体瘤细胞缺陷和下丘脑调控失常的发病机制曾提出过两种学说(垂体细胞自身缺陷学说和下丘脑调控失常学说)。现基本统一起来,认为垂体瘤的发展可分为起始阶段和促进阶段。在起始阶段,垂体细胞自身缺陷是起病的主要原因;在促进阶段,下丘脑调控失常等因素发挥了主要作用。即某一垂体细胞发生突变,导致癌基因激活和(或)抑癌基因失活。然后,在内外因素的促进下,单克隆突变细胞不断增殖,逐渐发展为垂体瘤。与其他肿瘤不同的是,垂体瘤常常伴有表观遗传学异常(epigenetic changes)。
癌基因诱导的细胞衰老机制紊乱
垂体微腺瘤(pituitary microadenomas)又称为隐性垂体瘤(occult pituitary tumors),垂体微腺瘤相当常见,但仅极少数发展为较大肿瘤,其原因是绝大部分微腺瘤在早期即出现生长静止(growth arrest),这一现象称为癌基因诱导的细胞衰老(oncogeneinduced cellular senescence,OIS),OIS起源于癌基因损伤,是组织抗增殖信号网络(antiproliferative signalling networks)活化的结果。因为正常人存在OIS机制,即使发生了微腺瘤,也可以被长期抑制而不增殖,但如果因为某种原因使OIS机制紊乱,则可由微腺瘤进展为大腺瘤。
细胞发生变异的原因为癌基因激活和(或)抑癌基因失活。现已查明的主要癌基因有c-myc、Rb、gsp、gip2、ras、hst及垂体瘤转型基因(pituitary tumor transforming gene,PTTG)等;抑癌基因有MEN-1、p53、Nm23及CDKN2A等。其中gsp基因在40%的GH瘤、10%的无功能腺瘤和6%的ACTH瘤中呈阳性。gsp基因及gip2基因激活使内源性GTP酶活性被抑制,于是Gs蛋白及Gi2蛋白α-亚基持续活化,后两者可分别看成是gsp癌基因和gip2癌基因的产物。这些癌基因产物可直接引起核转录因子如AP-1、CREB和Pit-1活化,使激素分泌增多并启动肿瘤生长。此外,癌基因激活导致胞内cAMP增加,后者刺激cyclin(细胞周期蛋白)D1和D3产生cdk2和cdk4,促进细胞由G1期进入S期。cAMP还诱导ras癌基因激活,ras癌基因与c-myc基因协同作用,阻止pRb与E2F结合,使细胞循环周期受阻,加快细胞由G1期进入S期。大多数垂体瘤组织缺乏CR6(正常垂体组织高表达),这可能是垂体瘤发病的因素之一。另一方面,多种垂体瘤的发病机制涉及抑癌基因 P16/CDKN2A的失活,该基因的CpG岛发生频繁甲基化是导致失活的重要原因。
但是,无功能性与功能性垂体瘤在肿瘤形成的本质上可能并无区别。临床上的无功能性垂体瘤均为单克隆性嫌色(嫌色细胞瘤)特征。在一般情况下,肿瘤分泌的LH/FSH不足以使血LH/FSH浓度升高,而血α亚基和铬粒素A(chromogranin A)增高。但在特定情况下,因肿瘤分泌的LH/FSH量较大而引起临床症状(LH/FSH瘤)。
