
促肾上腺皮质激素不敏感综合征(ACTH insensitivity syndrome)为遗传性疾病,包括家族性糖皮质激素缺乏综合征(familial glucocorticoid deficiency syndrome,FGD)和3A综合征两种不同的临床类型,临床以肾上腺皮质功能减退、皮肤色素沉着、皮质醇降低伴ACTH显著升高而盐皮质激素和RAS系统不受累为特征。
遗传性ACTH不敏感综合征包括3A综合征(triple A syndrome)和家族性糖皮质激素缺乏症(FGD)两种。临床表现不均一,同样的表现可由不同的遗传缺陷引起,而相同的遗传缺陷又可导致不同的临床表现。FGD的特点是仅有ACTH抵抗,其原因是ACTH受体(MC2R)基因或MC2R辅助蛋白(melanocortin 2 receptor accessory protein,MRAP)突变,而3A综合征还伴有许多其他临床特征,如低血糖症、生长发育障碍及神经功能异常等。
根据基因突变类型,FGD分为Ⅰ型和Ⅱ型两种。Ⅰ型是由于ACTH受体基因(黑色素皮质素2受体,MC2R)突变(约25%)或受体后缺陷所致。没有ACTH受体基因突变的FGD称为FGDⅡ型,由AAAS基因(定位于12q13,16个外显子)或MRAP基因(与ACTH受体的表达有关)突变引起,AAAS基因编码ALADIN(alacrina-achalasia-adrenal insufficiency-neurologic disorder)蛋白。少数FGD患者的病因仍不明了。
遗传缺陷导致ACTH不敏感
正常时,ACTH作用于肾上腺皮质,与ACTH受体结合后,以cAMP为第二信使而发挥作用。ACTH不敏感综合征可由于受体及受体后任何环节的缺陷引起。
现已发现5种黑色素皮质素(melanocortin)受体(MCR),分别命名为MC1R~MC5R,MC2R和MC5R由周围组织表达。MC1R即MSH受体,MC2R即ACTH受体。MC2R含297个氨基酸残基,属于G蛋白耦联受体(GPCR)家族中的吗啡样受体亚类。几乎仅在肾上腺皮质细胞膜表达。MC2R的基因无内含子,人ACTH受体与MSH受体的氨基酸/cDNA序列有39%完全相同,但与谷氨酸受体、PTH受体和降钙素(CT)受体的氨基酸序列没有同源性。ACTH受体只与ACTH结合。
ACTH受体突变
常染色体隐性遗传,ACTH受体缺陷有点突变、缺失和插入,以点突变居多:①第二穿膜区突变(如S74I)使ACTH受体与ACTH亲和力显著降低。②无义突变(如C201T)导致此密码子转变为终止密码子(TGA),使ACTH受体在第201位氨基酸以后(即羧基端胞膜内的第3袢以后)的肽链完全丧失,受体分子被截短。③C120G突变导致受体非极性化的第3穿膜区带正电荷,结合亲和力下降。
Allgrove综合征除有ACTH抵抗致糖皮质激素缺乏的临床表现外,还有眼泪缺乏、贲门失弛缓和进行性神经系统异常(包括自主神经功能紊乱),称为“3A”(adrenal insufficiency,alacrima,achalasia;AAA)或“4A”(adrenal insufficiency-alacrima-achalasia-autonomic disturbance)综合征。本综合征的临床特点是:①无泪症(alacrima):见于90%的病例,病因是泪腺萎缩;②贲门失弛缓症(achalasia of the cardia):见于85%的病例,病因是贲门神经元缺乏神经性一氧化氮合酶(endothelial nitric oxidase synthase,eNOS)和贲门肌层纤维化;③肾上腺功能不全(adrenal failure):表现为糖皮质激素缺乏(80%),15%患者伴有盐皮质激素缺乏。束状带和网状带萎缩,而球状带无损害。除以上表现外,患者多伴有进行性神经功能缺陷,如运动神经、感觉神经与自主神经病、智力减退、痴呆、视神经萎缩、共济失调、Parkinson病和感音性神经性耳聋等。
ACTH受体后缺陷
曾报告数例ACTH不敏感综合征患者的血ACTH升高,血皮质醇和去氢异雄酮降低,对外源性ACTH无反应。周围血单核细胞ACTH受体数目、受体与ACTH结合和G蛋白及其与腺苷环化酶的耦联均无异常,静脉滴注二丁酰cAMP无皮质醇和醛固酮升高反应;注射呋塞米或低盐饮食有醛固酮升高反应。3种肾上腺皮质激素(皮质醇、醛固酮和去氢异雄酮)均降低,提示患者对ACTH不敏感的缺陷在G蛋白或cAMP的下游,但缺陷的确切部位尚未确定。Yamamoto等报告两例(9岁和4岁)病例,血ACTH升高,但血皮质醇和尿17-OHCS正常。患者对外源性ACTH无皮质醇升高反应,低钠饮食刺激血浆肾素活性,醛固酮反应正常,血单核细胞ACTH受体对ACTH无反应,缺乏腺苷环化酶活性,受体的DNA序列正常,但对ACTH无高亲和力结合,提示既有受体与ACTH结合缺陷,又有受体后信号转导通路障碍。
其他遗传缺陷
Allgrove综合征的病因可能是丢失了与正常肾上腺和神经系统及皮肤角质化相连锁的必需基因。也有学者认为3A综合征是一个疾病谱,病谱的一端为单一性肾上腺皮质功能减退症,而另一端为贲门失弛缓症。病谱由各种重叠性缺陷组成,这些缺陷可累及骨骼系统、神经系统、皮肤、汗腺、内分泌系统、消化系统和免疫系统。患者出生时对ACTH反应正常,随后出现症状。Genin等通过染色体连锁扫描发现8q12.1-21.2区域有家族糖皮质激素缺乏综合征的候选基因。
MRAP突变导致家族性糖皮质激素缺乏症
Ⅱ型家族性糖皮质激素缺乏症表现为心理障碍、轻瘫及小脑畸形。病因与MC2R辅助蛋白(accessory protein,MRAP)突变有关。MRAP含单链穿膜肽段,位于细胞外的N-末端很长,最末端为可裂解的信号序列,而细胞内C-末端很短。与RTP、REEP比较,MRAP蛋白具有膜向性(membrane orientation),该3类受体无相同序列。MRAP蛋白促进靶细胞ACTH受体表达。正常MRAP蛋白位于细胞表面和内质网中,而突变型MRAP不能协助MC2R表达,故导致FGD(下图)。MRAP突变主要发生于其分子的第1编码区(外显子3)。但MRAP突变不能解释FGD患者的高身材,是否与受体活性修饰蛋白(receptor activity modifying protein,RAMP)有关仍未明了。患者表现为心理障碍、轻瘫、小脑畸形及高身材。血清皮质醇降低,而ACTH明显升高,且不被外源性皮质醇抑制。
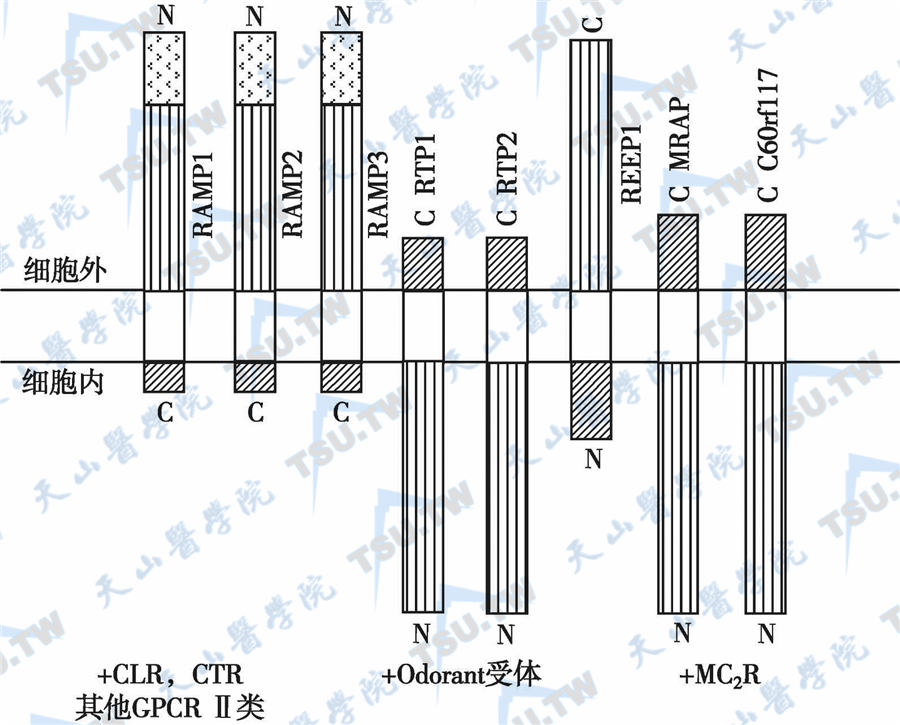
GPCR辅助蛋白家族的结构比较
注:MRAP:melancocortin 2 receptor accessory protein,黑皮素受体2辅助蛋白;RAMP:receptor activity modifying protein,受体活性修饰蛋白;RTP:receptor transporting protein,受体转运蛋白;REEP:receptor expression enhancing protein,受体表达增强蛋白;MC2R:melanocortin 2 receptor,黑皮素受体2(ACTH受体);N:N末端;C:C末端
