
英文:49,XXXXY syndrome。
溯源:1960年Fraccaro曾描述一例具有男性表型,但伴有睾丸小、阴茎小,先天性心脏病,枕部扁平,内眦赘皮和腹部膨隆的男孩病例。当时描述其核型是49,XXY,+18,+11。1962年进行染色体复查后,始确证其核型是49,XXXXY,而定名为49,XXXXY综合征。以往,常将本病例归入Klinefelter综合征范围。但是,随着本病病例数的增加和进一步的临床检查分析发现,本病虽具有与Klinefelter综合征相似的睾丸发育不全的典型症状,但却具有一些与Klinefelter综合征明显不同的临床特征,因而现已将本症确定为一种独立的综合征即49,XXXXY综合征。
发病机制
病因是男性核型中X染色体数增多。应用在X染色体上确定的Xq血型基因(定位于Xq),对相关X染色体进行Xq血型检查分析后,发现本综合征患者增多的X染色体源自母方,即患者母亲的卵细胞发生过程中,在第一次减数分裂和第二次减数分裂时,其X染色体连续发生不分离,因而形成具有26条染色体(22条常染色体和4条X染色体)的卵子,与正常精子(22+Y)受精后即形成49,XXXXY综合征患者。发病比例比49,XXXXX病例高10倍。
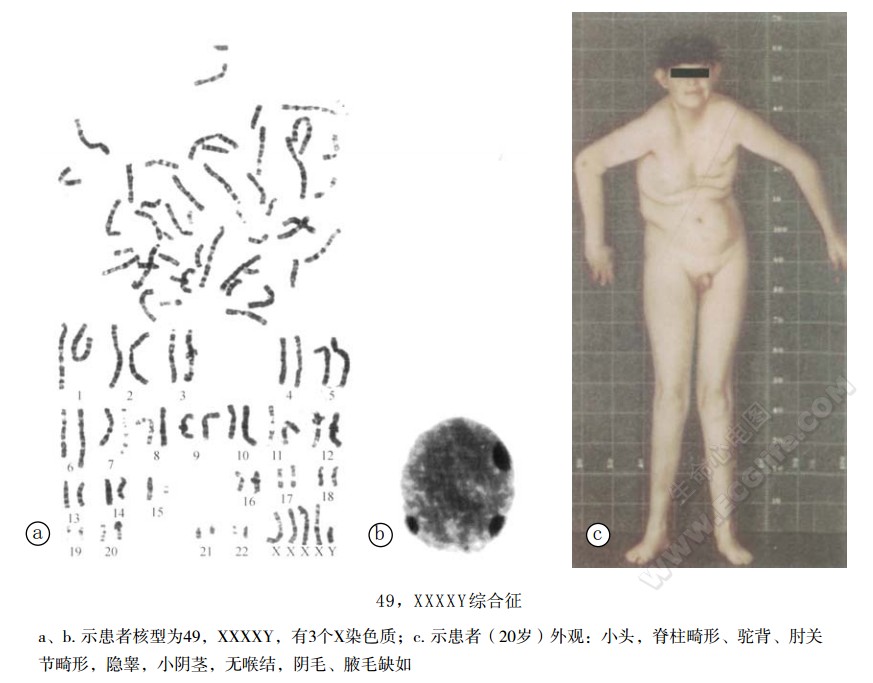
- 遗传学:常见的患者核型为49,XXXXY,X染色质3个,Y染色质1个,少数(约15%)为嵌合型。
- 皮纹学:指纹中总嵴纹数低。
临床表现
除具有与Klinefelter综合征类似的特征如外生殖器发育不全,曲细精管萎缩和玻璃样变性,缺乏精原细胞故不育外,本症尚具有不同的典型症状,如中度到重度智力低下(IQ:60~20),小头,眼距宽,内眦赘皮、斜视,鼻翼宽大。颈短,蹼颈,耳畸形、低位。脊柱后侧凸,驼背,肘关节畸形,此外,患者隐睾、睾丸小而硬,曲细精管中缺乏支持细胞,青春期尿中促性腺激素高等。
心血管损害:根据1975年Karsh统计,本综合征的3/4患者有先天性心脏病,最常见的是动脉导管未闭,其次为肺动脉狭窄和伴发肺动脉高压,房间隔缺损,冠状动脉异常等。
诊断
主要依据:
- 核型分析;
- 智力低下,小头,眼距宽,耳畸形、低位;
- 颈短,蹼颈,脊柱后侧凸、驼背,肘关节畸形;
- 隐睾,睾丸小而硬,阴茎短小;
- 心脏畸形。
预后
除伴发严重心脏畸形者外,一般可活至成年。
