
英文:Di George syndrome(DGS;MIM 188400);
同义名:先天性胸腺发育不良(congenital thymic hypoplasia),先天性胸腺不发育、胸腺发育不良综合征、先天性胸腺和甲状旁腺缺损综合征、Ⅲ~Ⅳ对咽囊发育不全综合征。
溯源与发展
1928年首由Marington报道1例出生后28天死亡的胸腺和甲状旁腺缺如病例。1956年儿科内分泌医生Di George将其于1963年见到1例先天性甲状旁腺不全的患儿,死后解剖发现无胸腺组织加以报道,并命名为先天性胸腺不发育。此后Huber、Taitz、Nahmias等都有病例报道,并称Di George综合征。我国于1978年董星祈等始有报道。其特征为特殊面容,新生儿手足搐搦,胸腺和憾甲状旁腺发育不良、缺如为特点的先天性免疫缺乏综合征。
发病机制
胚胎期第6~12周,第三和第四对咽囊开始发育形成胸腺、甲状旁腺和部分主动脉弓;同时鼻内侧突起融合为短人中,耳结节发育成外耳。如在此期间咽囊的发育受到干扰,就可导致上述器官和结构的缺损畸形。缺损可扩展至第一、第二对咽囊而累及面部结构,或累及第五对咽囊而致心脏异常。导致咽囊发育缺损的原因未明。近年来,一些作者认为Di George综合征(DGS)与圆锥干-面综合征(CTAF)具有共同的遗传基因,即22q11缺失,用半定量荧光PCR与多态性标记对两征分析,表明两者共同的易感区为22q11.2,说明不同临床心脏表型的CHD可有共同的遗传基因。
遗传学:呈AD方式遗传为多,但未最后确定。致病基因定位于22q11.2。
病理:可见胸腺和/或甲状旁腺发育不良,甚至缺如;细胞免疫系统不能正常分化,淋巴结皮质旁区即胸腺依赖区淋巴细胞数减少,网状内皮增殖和脾动脉周围淋巴结缺如;淋巴生发中心、骨髓和脾浆细胞则发育正常;心血管系统尤其主动脉弓发育畸形。1988年谈家桢对376例小儿尸解的胸腺病理学观察表明,在322例异常的胸腺组织中11例由原发缺陷引起,其中5例胸腺缺如,6例发育不良。
临床表现
由于胸腺缺陷,细胞免疫功能受损,患儿表现出对各种病毒和细菌的易感性。患者出生后3~4个月,即可出现风疹、水痘、慢性鼻炎、复发性肺炎、口腔念珠菌病、组织胞浆菌病、慢性腹泻和严重的结核感染。患儿接种牛痘、麻疹疫苗、卡介苗等,常可发生严重的致死性反应;输入含淋巴细胞的新鲜血液亦可发生致死性的移植反应。X线检查时看不见胸腺影。实验室检查发现患者细胞免疫功能低下,体液免疫功能如测定血免疫球蛋白等多正常,但亦有少数继发性体液免疫功能低下的报道。上述实验检查在新生儿期多正常,从婴儿期开始则明显异常。
新生儿期患儿即可有因甲状旁腺发育不全所致低钙抽搐。实验室检查可见低血钙、低尿钙、高血磷和血浆甲状旁腺激素浓度降低等。可因反复频繁抽搐、惊厥而致死亡。相当部分的病例可有面部畸形,如小头、宽眼距、内眦赘皮、眼裂下斜、小眼球、扁平鼻、短人中、小颌、唇或腭裂。耳低位、后旋和形态异常等。智力障碍、肝大、肺发育不良、肾缺失、食管闭锁不全等亦有报道。
心血管损害
85%~95%的病例有心血管畸形。心血管畸形中以大动脉畸形为最常见,占心血管畸形的95%左右,包括右位主动脉弓、永存动脉干、主动脉弓离断、大动脉转位等。可同时伴有法洛四联症、房或室间隔缺损、心内膜垫缺损、肺动脉狭窄、动脉导管未闭和两腔心等。心血管畸形是患者早期死亡的主要原因。
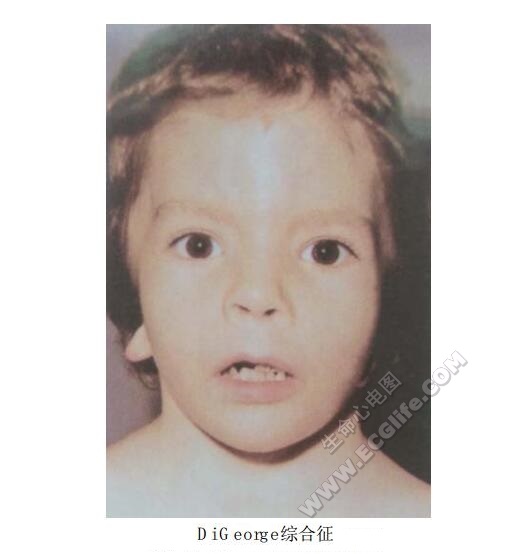
5岁女孩。新生儿期常发生低钙性抽搐。免疫功能差,生后3~4个月即反复发生呼吸道感染。小头,胸腺、甲状腺发育不良。智力低下,眼距宽,鼻孔朝天,人中长,耳低位、后旋,小下颌。右位动脉弓。
诊断
诊断主要依据胸腺及甲状旁腺缺陷的临床症状和实验室检查,加上心血管畸形或异常面容。Carrey认为胸腺缺陷、甲状旁腺功能不全和心血管畸形这三项主要诊断标准中,如无胸腺缺陷,则心血管畸形必须为主动脉弓异常,方能保证诊断的准确性。亦有作者认为依据患者的临床表现可将本病分为完全型和部分型两类。完全型者胸腺缺失或发育不良外,同时伴有甲状旁腺发育不良,心血管畸形和/或异常面容。部分型则可只有胸腺功能不全而不伴其他畸形。
治疗及预后
本病预后较差,长期存活少见。胸腺缺陷确诊后应尽早设法重建细胞免疫。治疗甲状旁腺功能不全可给予葡萄糖酸钙、大量维生素D、低磷饮食和甲状旁腺激素,以控制低血钙所致的抽搐和发育障碍。
心血管畸形主要采取手术治疗,早期手术常可改善患者的预后。
