
近年来随着生物化学、细胞生物学与分子生物学等学科的迅速发展,人们对血小板活化的过程和机制,特别是信号转导在血小板活化中的作用有了深入的认识。这对于血小板的生理病理变化以及血栓性疾病的防治都有重要的意义。
整合素αⅡbβ3(GPⅡb-Ⅲa)活化的信号转导
整合素αⅡbβ3是一个跨膜蛋白,其胞内段相对较短并且没有酶活性,αⅡbβ3主要通过结构和空间构象的修饰进行信号的转导。此外,αⅡbβ3具有变构效应的受体功能,可以促进跨膜的“双向”信号的转导。为与配体结合,整合素必须从对配体的低亲和力状态转变为高亲和力状态,这个过程即“内向外”信号转导。结合受体的整合素具有正受体功能,将细胞外环境的物理和生化信息传递到细胞内,并且调节细胞的迁移、繁殖、分化或生成,这个过程即“外向内”信号转导过程。“双向”信号转导在αⅡbβ3的胞内段主要依赖于β亚基的胞内段。循环中静息血小板其αⅡbβ3处于对配体的低亲和力状态,随着血管损伤和血小板激动剂如ADP、凝血酶、血栓素A2等的释放,血小板发生活化,这种活化过程中最重要的结果之一就是αⅡbβ3对配体如纤维蛋白原(Fg)的亲和力增高,相邻的αⅡbβ3因此发生交联从而导致血小板聚集和血栓形成。
许多胞内蛋白可与整合素胞内段结合,其中talin、α- actin、filamin、FAK等与大部分的整合素结合;而另一些胞内蛋白如CIB、Src、β3- endonexin6特异地与整合素受体αⅡbβ3相互作用。在这些分子中,整合素β亚基结合蛋白talin和Src特别受到瞩目。因为talin和整合素的结合是整合素活化过程中的最后通路;而在静息血小板中与整合素β3亚基结构性结合的Src激酶在“外向内”信号转导过程中对血小板功能也有重要的调节作用。目前认为,talin和整合素胞内段的相互作用诱导的构象改变通过αⅡbβ3的胞内段传递到胞外段,引起整合素受体对配基的由低到高的亲和力状态的改变。随着αⅡbβ3的活化,Fg与αⅡbβ3的结合引起αⅡbβ3构象进一步改变,这种变构信息通过胞外段β配基结合部位传递到细胞内段。在这种“外向内”信号转导过程中,Src通过磷酸化整合素β亚基胞内段以及其他具有酶活性的信号分子如局部黏着激酶FAK而发挥重要的调节作用。此外,信号转导过程中,talin的杆状区发生构象改变,隐藏的肌动蛋白结合蛋白vinculin的结合位点从而暴露,而局部聚集的vinculin与talin的结合增强了αⅡbβ3在肌动蛋白细胞骨架的锚定状态。但不同激活剂对于调节整合素αⅡbβ3功能的细胞内信号网络精细机制还不十分明确,目前一般认为主要通过以下3种信号转导机制调节αⅡbβ3的状态。
磷脂酶c(PLCβ)激活途径
磷脂酶c (PLCβ)激活途径是αⅡbβ3激活的经典途径(下图)。ADP、凝血酶、血栓素A2等与血小板上相应受体(P2Y1、PAR1或PAR4、TXR等)结合后,通过激活鸟嘌呤核苷酸结合蛋白(简称Gq蛋白)介导激活PLCβ,特异性水解磷脂酰肌醇(PIP2)产生第2信使三磷酸肌醇(IP3)和二酰甘油(DAG)。IP3动员细胞内Ca2+贮库,导致胞质内Ca2+浓度升高,促进了血小板整合素αⅡbβ3结合与纤维蛋白原受体的形成(integrin活化),并引起血小板的聚集。Amy E Brant等发现络合细胞内Ca2+或阻断Ca2+贮池入口均抑制了PLC诱导的αⅡbβ3的激活,进一步证明Ca2+是αⅡbβ3活化所必需的因素。DAG则可引起蛋白激酶C(PKC)的激活,后者通过磷酸化或去磷酸化β3亚单位胞质尾的丝/苏氨酸调节整合素αⅡbβ3的活化状态。
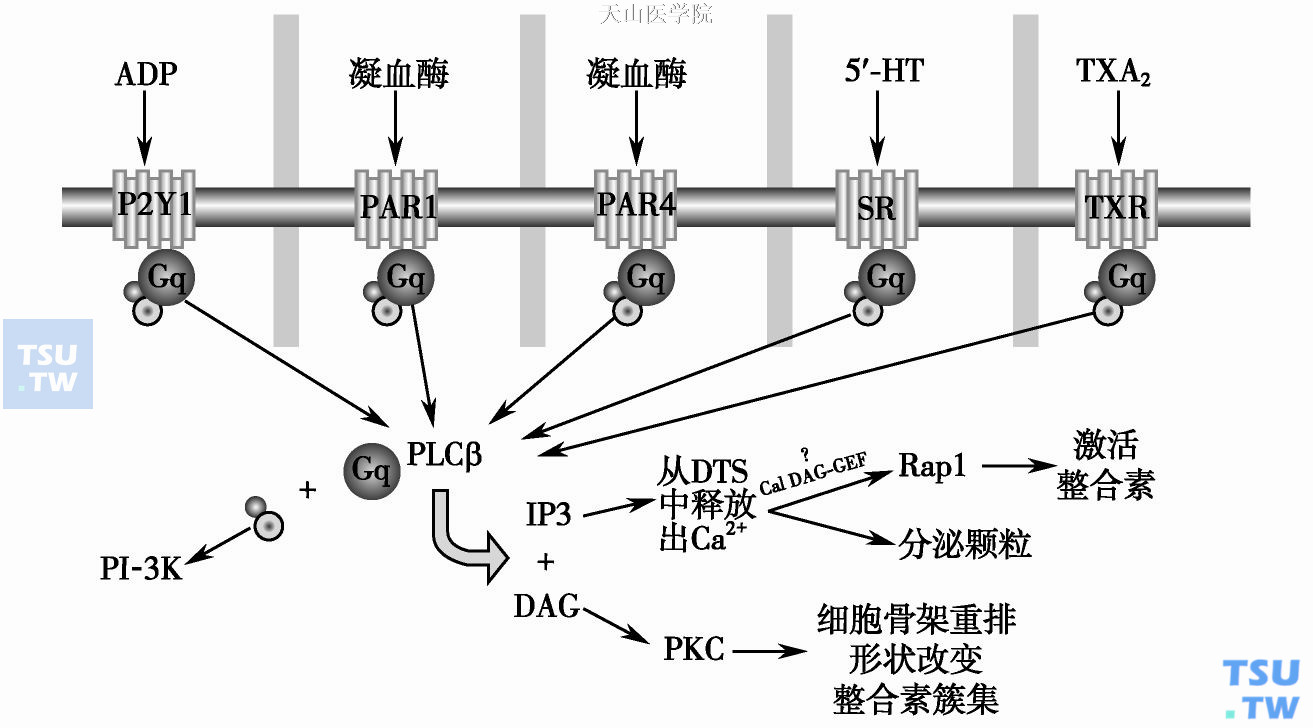
磷脂酶c(PLCβ)激活途径模式图
非受体酪氨酸激酶(如Src,Syk)激活途径
Fc受体(FcγRⅡA)胞质尾上含有一个免疫受体酪氨酸激活模序(ITAM)。当聚合的Ig与FcγRⅡA结合时,ITAM上的两个酪氨酸在非受体酪氨酸激酶Src家族激酶的作用下发生磷酸化,结合含有两个SH2结构域的Syk酪氨酸激酶和其他含有SH2结构域的蛋白质,引发Syk的激活,导致磷脂酶Cγ (PLCγ)、Btk、调节蛋白LAT、SLP276酪氨酸发生磷酸化而被激活,最终引起血小板的聚集。利用酪氨酸激酶抑制剂tyrphostin- A17抑制了凝血酶激活的整合素αⅡbβ3构象改变,肌动蛋白聚合,细胞骨架重构,并可部分阻断纤维蛋白原的结合;而用蛋白质酪氨酸磷酸酶抑制剂则可引起αⅡbβ3的活化,表明酪氨酸磷酸化和去磷酸化对αⅡbβ3激活至关重要。Fcγ或Syk基因敲除的小鼠中胶原诱导的血小板聚集功能缺失,表明胶原也通过同样的信号转导途径诱导血小板聚集。Hers等的研究表明Src参与αⅡbβ3的外向信号转导,最终是通过识别β3靶区域调节αⅡbβ3的活化状态的。
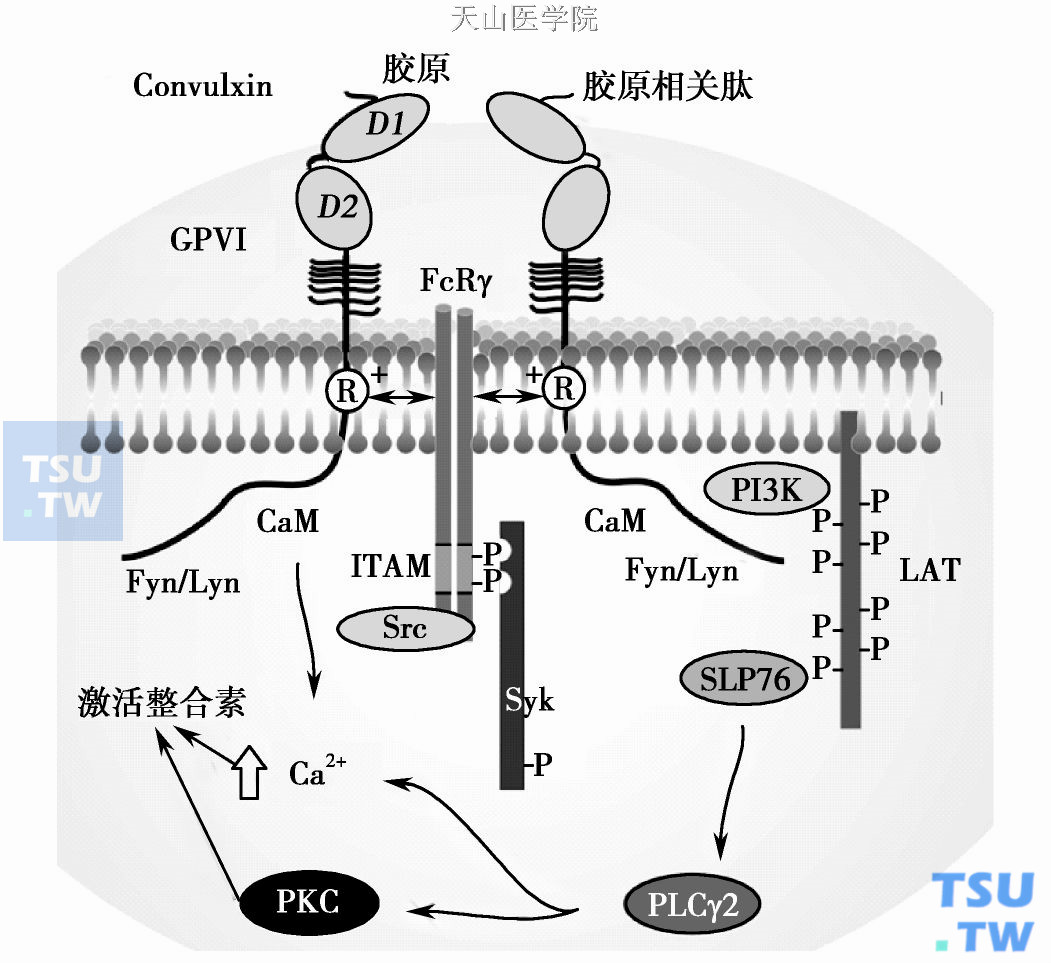
非受体酪氨酸激酶(如Src,Syk)激活途径模式图
腺苷酸环化酶激活途径
凝血酶、ADP与相应受体结合还可通过Gi蛋白抑制腺苷酸环化酶活性引起cAMP含量降低,抑制蛋白激酶A(PKA),引起αⅡbβ3活化。此外,cGMP含量降低引起的蛋白激酶G(PKG)活性降低也具有类似的功能,但具体机制还不清楚,只是实验证明小GTP酶Rap1B活化为该途径所必需。
GPⅠb-Ⅸ-Ⅴ活化的信号转导
GPⅠb-Ⅸ-Ⅴ复合物是血小板表面主要的黏附受体,是由GPⅠbα、GPⅠbβ、GPⅨ和GPⅤ四种亚基组成,能与许多种细胞外配体结合,包括vWF、凝血酶、P-选择素、FⅪ、FⅫ、Mac- 1。在高剪切力条件下(小动脉、粥样硬化病变的血管以及狭窄的血管等),血小板表面GPⅠb-Ⅸ-Ⅴ复合物与受损内皮下结合的vWF相互作用,激活产生一系列血小板活化转膜信号转导,最终导致整合素αⅡbβ3(GPⅡb/Ⅲa)活化和血小板聚集。在这一系列信号转导过程中,GPⅠb-Ⅸ-Ⅴ复合物与细胞内也有许多分子,包括肌动蛋白结合蛋白(ABP)、14- 3- 3ζ、钙调蛋白(CaM)等都发生相互作用,一起调节血小板的翻转、黏附、细胞骨架蛋白重排和细胞跨膜信号转导等血小板的功能。
ABP
ABP与GPⅠbα的细胞质尾部结合部位在氨基酸(aa)557~575残基,另一方面,ABP直接与肌动蛋白细丝结合,这样就能保证整个GPⅠb-Ⅸ-Ⅴ复合物锚定在细胞膜骨架上。GPⅠbα的胞质尾部与ABP相互作用对于高剪切力下血小板在基质vWF上黏附、翻转、活化和聚集都起着重要作用。
CaM
钙调蛋白(CaM)在静息血小板上与GPⅠb-Ⅸ-Ⅴ也有相互联系。CaM与GPⅠbβ的细胞质尾部的结合部位位于氨基酸(aa)149- 167残基,而与GPV相互作用的部位位于氨基酸(aa)529-544残基。当凝血酶刺激血小板后,CaM就从受体复合物上解离下来,而重新定位到细胞骨架上。虽然CaM与GPⅠb-Ⅸ-Ⅴ相互作用的生理作用还不是很清楚,但它们的相互作用可能在不同方面潜在地调节受体的功能和信号转导。
14- 3- 3ζ
14- 3- 3ζ是一个分子量为30kDa的血小板内到处存在的蛋白质分子。14- 3- 3ζ分子属于信号转导分子家族中一个成员,通过与丝氨酸磷酸化信号分子相互作用控制有丝分裂、细胞周期和凋亡等各种生理过程的信号转导。GPⅠb-Ⅸ-Ⅴ通过与14- 3- 3ζ的相互作用传递穿膜信号作用,细胞内Raf- 1、PKC和PI- 3K等许多各种蛋白都与14- 3- 3ζ有广泛的联系。在静息血小板内,14- 3- 3ζ与GPⅠb-Ⅸ-Ⅴ结合,而当血小板接受到固相化的vWF刺激后,14- 3- 3ζ就立即从GPⅠb-Ⅸ-Ⅴ解离出来,重新分布到细胞骨架上。14- 3- 3ζ在GPⅠb-Ⅸ-Ⅴ结合到vWF,诱导整合素αⅡbβ3活化,导致可溶性Fg与血小板结合和黏附细胞的伸展中的作用已在同时表达GPⅠb-Ⅸ-Ⅴ和αⅡbβ3的CHO细胞上得到证实。
14- 3- 3ζ在GPⅠb和GPⅤ上都有结合位点,14- 3- 3ζ 与GPⅠbα主要结合的位点位于GPⅠbα的胞质内C-端的4氨基酸,而且优先使GPⅠb尾部Ser609磷酸化,14- 3- 3ζ与GPⅠbα另外一个结合位点位于GPⅠbα中部aa 570- 590残基。14- 3- 3ζ与GPⅠbβ相互作用部位位于aa 160- 175残基,而且需要通过PKA对Ser166磷酸化来调节vWF结合到GPⅠb-Ⅸ。当GPⅠbβ发生S166A突变,或切割GPⅠbβ的Ser166部位时,就能增加vWF与GPⅠb的结合作用,所有这些表明14- 3- 3ζ与GPⅠbβ相互作用对于在高剪切力下GPⅠb-Ⅸ与固相状态下vWF的结合起着重要的负调控作用。14- 3- 3ζ与GPV的细胞质尾部结合部位在氨基酸(aa)529- 544残基。总之,14-3-3ζ通过组织信号复合物,包括PI- 3K和pp60SRC参与由GPⅠb-Ⅸ-Ⅴ诱导的血小板活化和聚集。
GPⅠb-Ⅸ-Ⅴ和磷脂酶C(PLC)
vWF结合到血小板GPⅠb-Ⅸ-Ⅴ引起PLC活化。当用瑞斯托霉素存在的vWF处理血小板,或在高剪切力下血小板黏附到固相化的vWF时,就引起细胞内Ca2+增加和PLC来源的第二信使IP3和DAG增加,从而引起血小板整合素αⅡbβ3的活化。PLCγ的酪氨酸磷酸化和PI- 3K的酯化产物(PIP2和PIP3)一起调节PLCγ的活化,而且PLC抑制物或者IP3受体拮抗剂能阻断血小板黏附到固相化的vWF上的形态改变。
14- 3- 3ζ和PI- 3K在GPⅠb-Ⅸ-Ⅴ信号转导作用
磷脂酰肌醇- 3激酶(PI- 3K)是一种磷脂酶,可分别催化4-磷脂酰肌醇,4,5-二磷脂酰肌醇3位羟基磷酸化,生成3,4-磷脂酰肌醇(PIP2)和3,4,5-二磷脂酰肌醇(PIP3)。血小板中含有两种PI- 3K异构体P85/P110,P110γ。P850/P110受酪氨酸磷酸化的蛋白质调控,而P110γ的活性主要由G蛋白的βγ亚基控制,与早期短暂的PIP2累积相关。PIP2和PIP3是跨膜转导信号,通过与蛋白质(如Akt,TIAM- 1或PLCγ,Src等)的PH结构域或SH2结构域结合,而将它们吸附至膜上。Jackson等应用wortmannin和LY294002等抑制PI- 3K进行的体外研究表明PI- 3K可调节较广范围的功能性血小板反应,包括最初的血小板黏附、细胞骨架重构、血小板的聚集等,但最重要的还是对GPⅡb/Ⅲa的亲和力的调节。
14- 3- 3ζ通过结合到PI- 3K完成活化的信号转导。在静息血小板中,PI- 3K通过14- 3- 3ζ与GPⅠb-Ⅸ-Ⅴ相互联系,血小板经vWF活化后,PI- 3K转位到细胞骨架与GPⅠb-Ⅸ-Ⅴ和14- 3- 3ζ联系。血小板在有瑞斯托霉素存在的vWF刺激时,或者在血小板黏附到固相化的vWF时,PI- 3K也被活化。在高剪切力条件下,PI- 3K与酪氨酸激酶Syk相联系;而且,在vWF刺激后的血小板,PI- 3K通过Src的SH3区和PI- 3K的调节亚基p85相互作用与GPⅠb-Ⅸ-Ⅴ和Src形成复合物,从而通过酪氨酸磷酸化和PI-3K的脂化产物(PIP2和PIP3)诱导PLCγ2活化,最终激活整合素αⅡbβ3的活化,导致血小板聚集。
cGMP-依赖的蛋白激酶
一般认为cGMP和cGMP依赖的蛋白激酶(PKG)水平增高抑制血小板的活化。近来,研究显示cGMP- PKG途径在GPⅠb-Ⅸ-Ⅴ调节血小板活化信号转导中起着重要的激活作用。Li Z等在CHO123(同时重组表达GPⅠb-Ⅸ和GPⅡb-Ⅲa)细胞上重组表达PKG的细胞(PKG- CHO123)模型研究表明vWF结合到GPⅠb-Ⅸ后诱导PKG和cGMP的表达增高,激活整合素αⅡbβ3的活化,PKG- CHO123细胞结合Fg能力显著增高;而且重组表达PKG- CHO123细胞在固相化vWF表面上稳定黏附能力显著高于CHO123细胞,另外cGMP能增强PKG- CHO123细胞的黏附,而RGDS多肽能抑制PKG- CHO123的黏附。PKG敲除小鼠的血小板在vWF表面上伸展能力显著降低,而且PKG水平受cGMP的反馈调节。但也有一些相反报道,vWF不能增加cGMP水平,而且增高cGMP反而抑制vWF/GPⅠb-Ⅸ-Ⅴ相互作用引起的血小板活化。
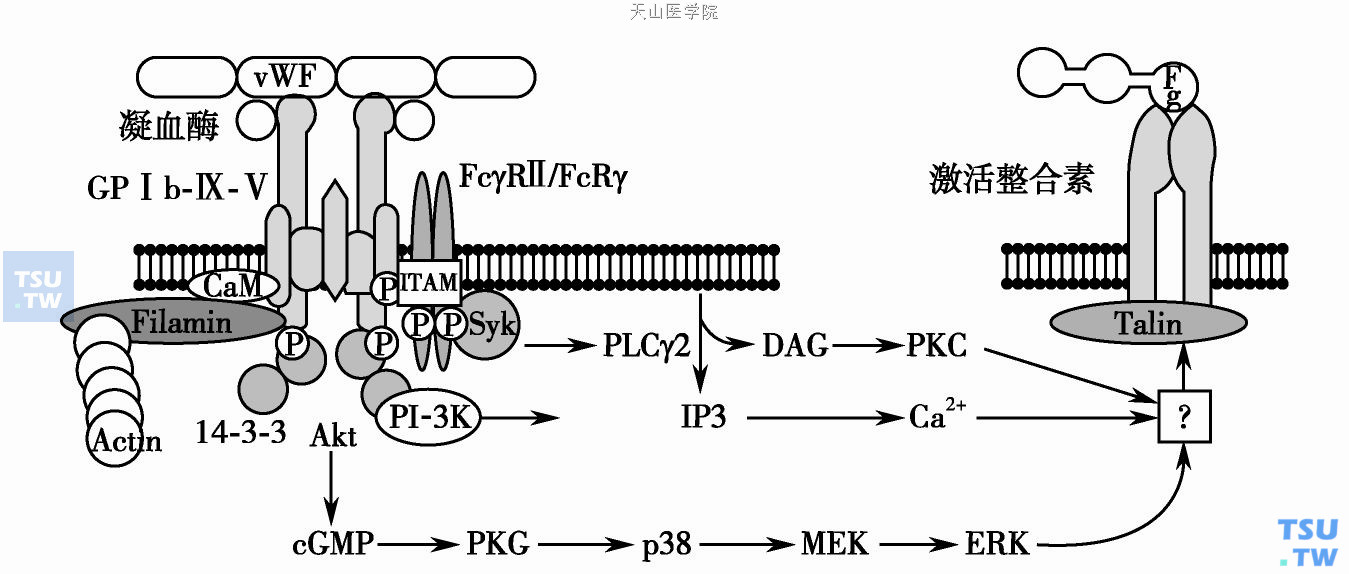
cGMP依赖的蛋白激酶在GPⅠb-Ⅸ-Ⅴ活化的信号转导中作用
总之,GPⅠb-Ⅸ-Ⅴ和整合素αⅡbβ3的耦合和协同效应在血小板黏附到内皮下胶原和血栓形成中起着关键的作用。在静止血小板上,整合素αⅡbβ3对可溶性的配体表现出非常低的亲和力。然而,在高剪切力条件下,血小板通过GPⅠb-Ⅸ-Ⅴ结合到基质vWF,血小板启动“内-外”信号转导,将整合素αⅡbβ3转换成高亲和力形式,从而使其与各种配体有效结合,另一方面血小板整合素αⅡbβ3的“外-内”信号转导引起血小板的第二次活化反应,促进α颗粒与致密颗粒的释放,使更多血小板活化和聚集。同时血小板膜磷脂的翻转促进了血液的凝固过程,而某些α颗粒成分(PDGF、TGF- B、PF4等)的释放会影响血管的生长与功能,P-选择素与PF4又参与其炎症反应。在静止或流动条件下,由GPⅠb-Ⅸ-Ⅴ诱导的活化存在着许多活化信号途径引起整合素αⅡbβ3的活化,而起着最重要和中心作用的是细胞内Ca2+。在不同的实验条件下,GPⅠb-Ⅸ-Ⅴ可以通过启动不同的信号途径引起整合素αⅡbβ3活化。虽然GPⅠb-Ⅸ-Ⅴ和整合素αⅡbβ3活化的信号转导是一个复杂的过程,对于其结构功能及活化本质的深入研究无疑有助于人们深入认识血小板黏附、聚集、血栓形成的机制及血栓性疾病的发病机制,寻找血栓性疾病诊断和抗血小板治疗和抗血栓治疗的新的靶点。
(赵益明 阮长耿)
