
半乳糖血症(galactosemia)是一种遗传性糖代谢缺陷症,由于乳糖或半乳糖转变为6-磷酸葡萄糖所需的酶活性缺如或明显减低,半乳糖转变为6-磷酸葡萄糖发生障碍而使半乳糖及其旁路产物在组织和细胞堆积,影响器官、组织和细胞的功能,临床以肝功能衰竭、中枢神经系统退行性变、白内障和女性高促性腺激素性卵巢早期衰竭为特征。本病为常染色体隐性遗传,不同地区、不同民族人群中的发病率存在着差异,全世界均有本病的病例报道。
半乳糖代谢需要四种酶催化
β-D-半乳糖转化为1-磷酸葡萄糖需要4种酶的参与(Leloir途径,下图)。在图中共有4种酶,共同组成Leloir通路。在代谢过程中所产生的1-磷酸葡萄糖,还要经转位酶作用转变为6-磷酸葡萄糖再进入葡萄糖代谢循环。引起遗传性半乳糖血症的酶缺陷主要是GALK、GALT和GALE,由于酶缺陷而使半乳糖主要代谢通路受阻,除引起半乳糖-1-磷酸和半乳糖在组织和细胞中堆积,还导致半乳糖代谢旁路开放,产生有害的半乳糖醇(galactitol)和半乳糖酸(galactonate)。
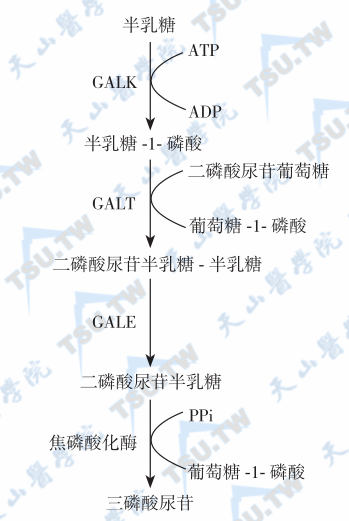
半乳糖的主要代谢途径
注:GALK:半乳糖激酶;GALT:半乳糖-1-磷酸尿苷转移酶;GALE:尿苷二磷酸半乳糖-4-差向酶
上述3种酶由不同基因编码,GALK、GALT和GALE基因分别定位于9p13、17q24和1p36,GALK长度为7.3kb,与胞质内浆网膜相连系,共8个外显子;GALT长4kb,11个外显子,人、小鼠、酵母和大肠杆菌的相同的酶,在外显子6、9和10的一部分有相同的保留序列;GALE长4kb,含11个外显子。3种酶的等位基因均存在不均一性,如GALT的等位基因即存在D/G、G/G、D/Los、Angeles等。等位基因又有D1和D2两种,前者增加酶活性,后者恰恰相反(D=duarte)。GALK是乳酸代谢过程中的第一步所需的酶,属小分子质量激酶,属GHMP(galactokinase,homoserine kinase mevalonate kinase and phosphomevalonate kinase)。已发现GALK基因的20多种突变,主要为缺失和点突变,其中P28T为罗马人奠基性突变。突变所致影响只限于晶状体而不牵涉全身。
GALE酶缺陷影响半乳糖代谢的第三步。GALE基因也有许多突变,但突变对酶活性的影响因突变不同而异,有的只牵涉周围某些组织如晶状体,有的则影响全身。前者称为“良性”周围型半乳糖血症;后者为严重型半乳糖血症,其临床表现与GAKT酶活性完全丧失相似,如V94M和G90E等。同样引起半乳糖的堆积,为什么GALE有些突变只影响局部,有些突变则引起全身受累,目前尚无法解释。
相关代谢酶突变导致半乳糖血症
经典的半乳糖血症为常染色体遗传性疾病,病因为半乳糖代谢过程中所需的3种酶基因发生突变,使所表达的酶活性全部或部分丧失。到目前为止,已有229种序列异常,其中突变类型196种。以GALT为例,等位基因为G/G型,则酶活性全部消失;D/G型则酶活性丧失一部分。迄今为止GALT已被发现有160多种突变,包括错义、无义、移码、拼接和缺失突变,在不同种族人群中每种突变的频率不相同,以Q188R最为常见,K285N次之。
