
英文:acrocephalosyndactyly type Ⅰ syndrome;
同义名:尖头并指(趾)综合征Ⅰ型、并指型尖头综合征、Apert综合征。
溯源与发展
1906年首由Apert报道9例,以后得名Apert综合征。1920年Park和Powers又报道20例并详加描述,认为是一个独立的综合征。1960年Buckley指出此并指为骨性融合,多见于2~4并指。我国于1966年黄少勇等开始报道,目前已有少数病例报道。Blank及高桥认为,本征是常染色显性遗传,由冠状缝过早闭合而形成的尖头、颅骨、耳骨发育畸形、伴并指(趾)为特征的综合征。
发病机制
目前认为是一种遗传性先天性畸形,Blank指出在高龄夫妇新生儿中发生率高,尤其是与生育年龄过高有重要关系,认为老年机体代谢产物是一种化学异常刺激物,而导致双亲生殖细胞发生基因突变,由于基因多效性,而产生多系统发育缺陷。
遗传学:呈AD方式,多数为新发生突变,致病基因定位于10q26。
临床表现
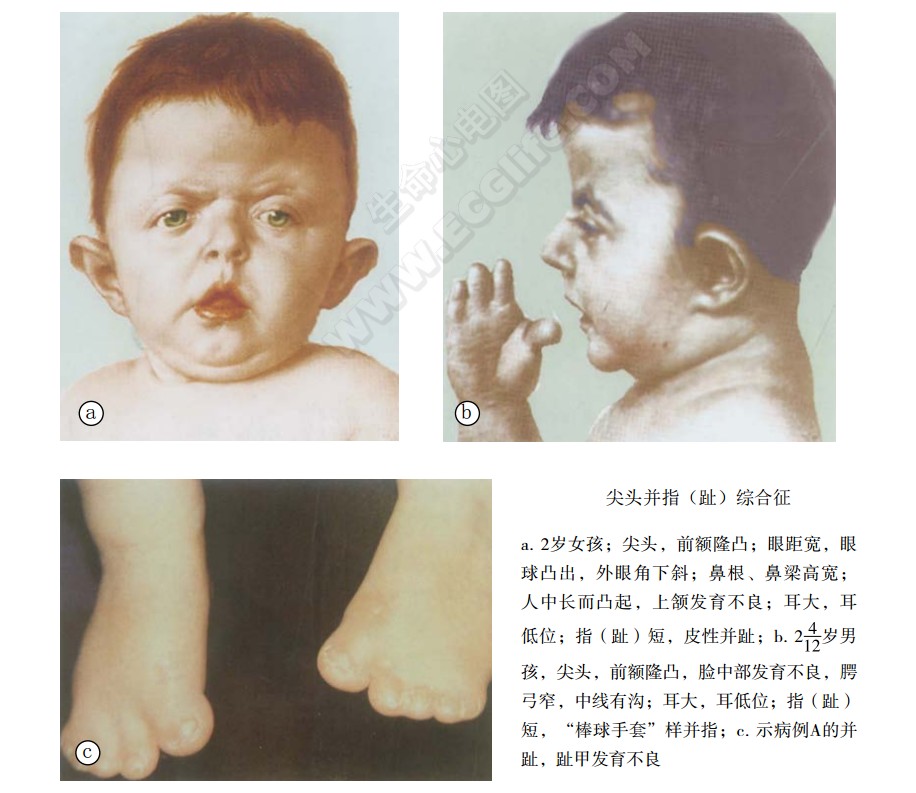
各骨缝间早闭,尤其是冠状缝融合导致大囟门向上隆起,颅顶短而高耸,呈塔形尖头,枕骨扁平,颜面骨形成异常,眼上缘发育不全,形成眼眶浅,眼球突出,眼睑裂缝向下或向外倾斜,眼距宽,斜视,鼻短而呈钩形,上颌发育不全,下颌突出,面部不对称,高腭弓,伴正中沟、软腭裂、悬雍垂裂,牙齿拥挤,牙骀错位,呈特殊面容。皮肤遍生痤疮,智力正常或迟钝,听力丧失,手足骨或软组织的并指(趾)畸形,最常见累及第2~5指(趾),指(趾)关节强直,肘关节强直,指甲畸形,严重者有内脏器官畸形。
心血管损害:发生率约为10%。常见的为室间隔缺损、法洛四联症、主动脉缩窄和传导阻滞等改变。
鉴别诊断
患者具有典型的面貌,颅面骨形成异常,伴有并指(趾)畸形,即可考虑诊断,如有AD遗传家族史,更有助于确诊。
需与下述疾病相鉴别:
- 尖头并指(趾)畸形Ⅱ(MIM 201000,肥胖,性功能低下);
- 尖头并指(趾)畸形Ⅲ型(MIM 101400.头颅不对称,经度并指(趾));
- 尖头并指(趾)畸形V型(MIM101600,拇指宽而短,脚趾大,2、3趾并趾)。
治疗
对颅骨早期愈合者,应行颅骨减压术,防止视力和智力低下发生和发展,并指和心血管畸形者可行矫正手术,美容治疗可改善面貌异常,口腔治疗也是非常重要的,如出现精神迟缓,特殊教育可有所帮助。
