单纯性肥胖(simple obesity)的病因和发病机制尚不完全清楚,其主要原因是摄入的能量大于消耗的能量,但遗传因素不可忽视。脂肪细胞来源于成纤维细胞的分化。正常成人约含有350亿个脂肪细胞,每个脂肪细胞含0.4~0.6μg甘油三酯。重度肥胖者的脂肪细胞数目可增加至正常的4倍,而每个脂肪细胞的含脂量也相应加倍,这样一来,重度肥胖者的体脂含量可达到正常人的10倍。肥胖者体内过多的脂肪具有浸润作用,导致脂肪肝、血脂谱异常、糖尿病和动脉粥样硬化等。
一般认为,人类的种族易患性、肥胖基因和肥胖相关基因变异(突变与多态性)以及个体的代谢类型(食欲、消化吸收功能、睡眠质量和代谢效能)是单纯性肥胖的发病基础,而不良生活方式(体力活动过少和能量摄入过多)为发病的必要条件。流行病学调查表明,多数单纯性肥胖者有家庭发病倾向。肥胖父母所生子女中,患单纯性肥胖者比父母双方体重正常者所生子女高5~8倍;但多数单纯性肥胖并非肥胖基因或肥胖相关基因变异所致。从大样本肥胖人群的调查中发现,约有250个基因或表达序列标志(expressed sequence tag,EST)的功能与肥胖有关,其中有些基因的生物学行为(biologic behavior)可能在肥胖的发病中起了关键作用(主效基因,master genes),而另一些基因所起的作用相对较弱,但目前的研究还不深入。
基因变异导致肥胖
单基因突变所致肥胖的特点是具有明确的遗传性,肥胖发生年龄早、进展快、肥胖程度重和并发症多。
肥胖基因突变
肥胖(ob)基因位于第6号染色体上,与Pax4非常接近,同时紧靠一限制性片段长度多态性标志D6RCK13。肥胖基因由3个外显子和2个内含子组成,编码4.5kb mRNA,其表达产物为瘦素,由外显子2和3编码。瘦素的mRNA含有167个氨基酸残基组成的开放性阅读框架。瘦素由白色脂肪组织分泌,其分泌呈脉冲式,并具有昼夜节律。瘦素通过与其受体(有4种异构受体)结合而发挥其生理作用。将体内脂肪贮存的信息传送到下丘脑和弓状核饱食中枢,减少神经肽Y的分泌,摄食减少。ob/ob小鼠有多食、肥胖、高血糖、高胰岛素血症、糖尿病、低体温和不育;而db/db小鼠的表型虽与ob/ob相同,但血瘦素水平升高。将db/db小鼠与野生型小鼠联体共生,则可使野生型小鼠的摄食减少而致死。由此可见,瘦素与调节摄食及肥胖的发生有关。人的瘦素基因突变可引起极度肥胖。此外,瘦素基因突变还与低促性腺激素性性腺功能减退症、免疫功能异常、高胰岛素血症相关,并与儿童生长发育迟缓、继发性甲状腺功能减低等亦有一定的病因关系。
其他基因突变
POMC基因突变可能与肥胖和肾上腺皮质功能减退有关。激素原转换酶1(prohormone convertase 1)基因、黑皮素4受体(melanocortin 4 receptor,MC4)基因、激素原转换酶1和SIM1基因突变可引起肥胖。近来发现,kisspeptin具有多种生理作用,其中最主要的是调节生殖和性激素分泌,kisspeptin是联系营养和生殖功能的物质基础,可能与肥胖有重要联系。单基因突变的表型及其与人类肥胖的关系见下表。
单基因突变的表型及其与人类肥胖的关系
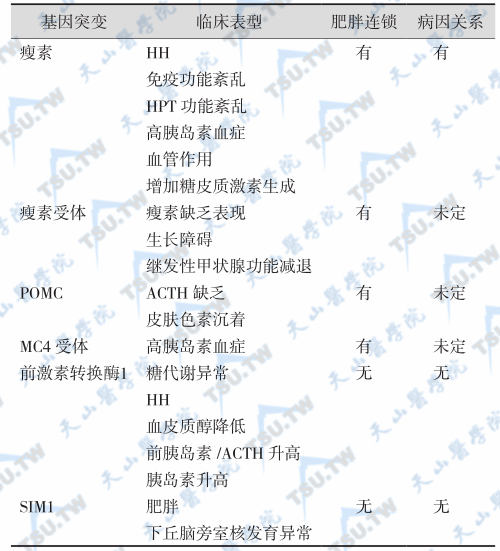
注:HH:hypogonadotropic hypogonadism,低促性腺激素性性腺功能减退症;HPT:hypothalamus-pituitary-thyroid axis,下丘脑-垂体-甲状腺轴;POMC:pro-opiomelanocortin;MC4:melanocortin-4。
精神心理因素刺激摄食行为
刺激下丘脑的腹内侧核可使动物拒食,而完全破坏这一神经核则引起多食。周围神经系统对摄食也有调节作用。神经肽的食欲兴奋性(orexigenic)和食欲抑制性(anorexigenic)信号分别通过各自的受体途径而影响和调节机体的食欲与食量;进食足量后,通过周围神经将“饱感”信号传送到中枢神经,因而停止继续进食。神经精神方面的任何异常均可通过心理应激、精神感觉和运动功能的改变而促进食欲,导致肥胖。在悲伤或过于兴奋的情况下进食减少,说明精神因素对摄食也有调节作用。在临床上,下丘脑病变易引起肥胖或消瘦。神经性贪食患者具有极度饥饿感和贪婪的食欲,患者要满足饥饿感就不停地进食,通常暴饮暴食,暴食后又引吐,这种现象与精神压抑和强迫观念有一定关系,但具体的发病机制未明。
Facchinetti等在13名肥胖儿童中发现,血浆β-内啡肽升高,且不能被地塞米松抑制,推论肥胖儿童的β-内啡肽不受CRH的控制,而阿片类拮抗剂纳洛酮可使多食现象消失。肥胖者有胰岛素抵抗和高胰岛素血症,后者引起胰岛素受体降调节,又进一步增加胰岛素抵抗,形成恶性循环。胰岛素分泌增多,刺激摄食,同时抑制脂肪分解,因此引起体内脂肪堆积和肥胖。脂肪组织中的酶活性升高是发生胰岛素抵抗的重要原因。
某些激素促进食欲并诱发肥胖
调节摄食行为的激素很多,其中较肯定而明显的激素主要是皮质醇、雌激素与瘦素。
皮质醇
单纯性肥胖者的皮质醇生成量增多,但因组织对皮质醇的清除增加,故血清皮质醇不一定升高。脂肪细胞在11β羟类固醇脱氢酶的作用下生成皮质醇,而且皮质醇的生成量与脂肪细胞的数量呈正比,因此可出现Cushing综合征样体脂分布和中心性肥胖。
雌激素
青春期开始时,体脂约占体重的20%。男性在青春期末的体脂减少到15%,而女性则增加到25%,成年肥胖以女性居多(特别是经产妇和口服避孕药者),提示性激素在单纯性肥胖的发病中起了一定作用。女性的体脂比例高于男性,而且其体脂的分布特殊(女性体脂分布和女性体型),绝经后体脂重新分布,多余的体脂同样积聚于内脏,故绝经后肥胖女性的心血管病和T2DM的危险性较绝经前明显增加,说明雌激素起了重要作用。体外试验发现,雌激素对11β羟类固醇脱氢酶的影响具有组织特异性,雌激素降低该酶在肝、肾和睾丸的活性,但升高内脏组织前脂肪细胞的活性。因此,雌激素可增加皮下脂肪细胞的体积,抑制脂解;而绝经后因雌激素缺乏使脂解增加,PAI-1减低,心血管病风险增加。
食欲素与瘦素
食欲素可增强食欲,饥饿状态可上调前食欲素原表达。食欲素A受体(OX1R)属于G蛋白耦联受体家族成员的一种,食欲素B受体(OX2R)与OX1R有64%的序列同源,两种受体存在交叉结合现象。OX1R和OX2R仅存在于脑组织中,主要分布于下丘脑的“摄食中枢”,而瘦素受体主要分布于“饱食中枢”。
瘦素是重要的能量调节激素。肥胖和代谢综合征患者的高胰岛素血症、胰岛素抵抗、免疫功能异常等均与瘦素抵抗有关。中枢性瘦素缺乏综合征(central leptin insufficiency syndrome)是指下丘脑和其他脑细胞缺乏瘦素活性,导致能量代谢调节障碍;瘦素抵抗综合征(leptin resistance syndrome)通过刺激脑组织的瘦素受体和抑制食欲而降低体重,但单独用外源性瘦素并不能减低肥胖者的体重,因为肥胖者并不缺乏瘦素,相反存在瘦素抵抗。肥胖者脂肪细胞分泌的瘦素增多,后者作用于下丘脑的瘦素受体,抑制神经肽Y(NPY)的分泌并促进α-MSH的释放,α-MSH作用于MC4(摄食抑制性)受体,抑制食欲。瘦素也抑制agouti相关肽(AGRP,α-MSH拮抗剂)的分泌,使摄食减少,体重下降。中枢神经系统存在促进食欲和抑制食欲与摄食行为的两套调节系统。神经肽Y、黑色素浓集素(melanin concentrating hormone,MCH)、食欲素A和B、甘丙素及agouti相关蛋白均为促进食欲的调节因子,而α-MSH、CRH、胆囊收缩素(CCK)、可卡因和苯丙胺调节性转录物(cocaine and amphetamine regulated transcript,CAR)、神经降压素(neurotensin)、胰高血糖素样肽-1(GLP-1)和蛙皮素均为抑制食欲的调节因子。
能量摄入过多引起肥胖
不爱活动的人能量消耗减少,易发生肥胖。运动员在停止运动后、经常摄入高热卡饮食、睡前进食或吸烟者在戒烟后都与单纯性肥胖的发生有关。能量摄入和能量消耗之间的平衡反映在体重上。
节俭基因型
近几十年的人类生存环境发生了巨变,这种变化远远超越了人类进化的速度和对环境的适应能力,人类的体重基本上缺乏有力的调节机制,人类生存环境的巨变必然影响到基因的表达和功能。环境巨变远远超越了人类的进化速度和对环境的适应力,环境因素通过“节俭基因型”(thrifty genotype)和“共同土壤(common soil)”导致肥胖。人类生存的环境的巨变必然影响到基因的表达和功能。另一方面,现代文明显著减轻了体力活动的负担和能量消耗。
人类进化过程中所选择的“节俭”基因有利于食物充足时促进脂肪堆积和能量储存,以供天灾饥荒时食物短缺时耗用。因此,具有在进食后能较多地将食物能量以脂肪形式储存起来的个体,就较易耐受长期饥饿而生存下来。这种有“节俭”基因型的个体在人类进化中有利于在逆境中生存而被保留下来。但是到了食品供应充足的现代社会,有“节俭”基因的个体就易出现肥胖、胰岛素抵抗和糖尿病;也就是说,在体力活动减少、热量供应充足的情况下,“节俭”基因转变成了肥胖和T2DM的易感基因。流行病学调查表明,糖尿病、高血压、血脂紊乱、肥胖等这些成人常见病在家族中有聚集现象(代谢综合征)。“共同土壤”假设认为,这些疾病有各自不同的遗传和环境因素参与发病,但还可能有共同的遗传及环境因素基础,家族内孪生子、同胞及亲属患者之间上述并发症发生的一致率高。
能量摄入过多
能量消耗的去路有静息性能量消耗(resting energy expenditure)、热量生成(themogenesis)和体力活动(physical activity)。静息性能量消耗由个体的大小和机体成分等因素确定,一般占能量消耗总量的50%~80%;热量生成用于食物的消化、吸收和体温的调节,约占10%;静息性能量消耗和热量生成是基本固定的,而体力活动所需的能量差异很大。但是,人类能量摄入和能量消耗之间的平衡主要靠个体的主观感受和行为自我控制。摄食行为容易受许多特殊食物、环境因素和心理因素的刺激,引起摄食过多。因此,个体每天的能量摄入量差异平均波动在20%~40%,而体力活动的波动更大。
20世纪30年代,有人把一群小鼠随机分成两组,一组为限量组,喂食量为正常量的60%;另一组可以自由进食。1000天后,限量组小鼠的骨骼还在缓慢发育生长,平均存活1300天;而对照组小鼠6个月后骨骼全部停止生长,平均寿命仅900天,而且肥胖与肿瘤的发生率也比限量进食组高得多。这就是所谓的“麦卡效应”。以后的动物实验也得出相近的结论。有人曾做过另1项动物实验:两群猴子,一群吃饱为止,一群的进食量仅七八分饱。10年后,每餐吃饱的猴子腹部膨大,患血脂紊乱、脂肪肝、冠心病多,100只猴子只有50只存活。另一群猴子健康,精力充沛,100只中存活了88只。15年时,每顿饱餐的猴子全部死亡,高寿的猴子都在进食较少的群落中。
能量密度过高
能量密度是指食物中脂肪的含量和比例,食物中的脂肪含量和比例越高,其能量密度(energy density)也越大。能量密度在人类食欲和能量摄入行为的调节中起了重要作用。现代食品工业尽力提供高甜度高能量食品,以适应人们口感需要。现代饮食的另一个问题是高脂肪。人们被脂肪的香味所诱惑,食物的能量密度相当高。
代谢效能过强
机体将体外能量物质转化为自身贮存能量的效率差异很大,这种差异可理解为代谢效能(metabolic efficacy)。胖者和瘦者的Na+/K+-ATP酶活性和对各种激素及环境刺激的代谢效能是不一样的,β3-肾上腺能受体在肥胖的病因中有重要影响,可认为它是一种肥胖候选基因。静息代谢率(resting metabolic rate,RMR)的个体差异主要由机体中的瘦体质(fat-free mass)和遗传因素决定,此外也受甲状腺激素水平、交感神经活动性等的影响。RMR似乎是肥胖“易感因素”中最重要者。老年人往往因胰岛素抵抗和体力活动减少而导致肥胖,其中肌肉组织的胰岛素抵抗还伴有细胞线粒体功能紊乱,心肌GLUT4和解耦联蛋白-3表达降低,代谢效能明显降低,因此更易引起肥胖。
糖皮质激素过多引起Cushing综合征,包括了代谢综合征的所有成分,如肥胖、T2DM、高血压、血脂紊乱、心血管病变等。在代谢综合征和肥胖中,虽然血清糖皮质激素水平不高或稍微升高,但更突出的表现在脂肪组织的低度炎症(low-grade inflammation)与1型11-β羟类固醇脱氢酶(11beta-hydroxysteroid dehydrogenase type 1,11β-HSD1,基因HSD11B1)活性升高。11β-HSD1反映了糖皮质激素在细胞内的作用强度,其活性越高,引起的炎症反应和能量-物质代谢的效应也越大。
慢性炎症
慢性炎症与肥胖(如进食行为异常)的关系密切,炎症还是许多肥胖并发症(如血管病变)的主要原因。但是目前对两者的联系机制了解甚少。
不安全食物
肥胖与不安全食物(food insecurity)亦有一定关系。不安全食物引起肥胖的原因是多方面的,可能主要与人为地增加食物的美感、色泽、含糖量、调味剂、食欲促进剂等有关,而要达到此目的,就很可能需要添加一些不安全的物质。
疾病和药物促发肥胖
疾病导致的肥胖
疾病和药物促发的脂肪堆积属于继发性肥胖的范畴,但对理解肥胖的发病机制很有帮助。神经精神疾病、下丘脑疾病、Cushing综合征、慢性酒精中毒是继发性肥胖的常见原因,这类疾病的共同特点是下丘脑功能紊乱,可能通过摄食、食欲和其他一些未知因素促进了肥胖的发生与发展。此外,进行腹膜透析患者易发生肥胖,而肥胖又促进肾功能恶化。流行病学资料显示,患过先兆子痫的妇女以后易发生心血管病,因先兆子痫常与糖尿病、高血压、血脂紊乱、肥胖和代谢综合征相联系。研究表明,母乳喂养可在一定程度上预防肥胖的发生,此可能与母乳含有一些特殊的营养成分有关。
药物导致的肥胖
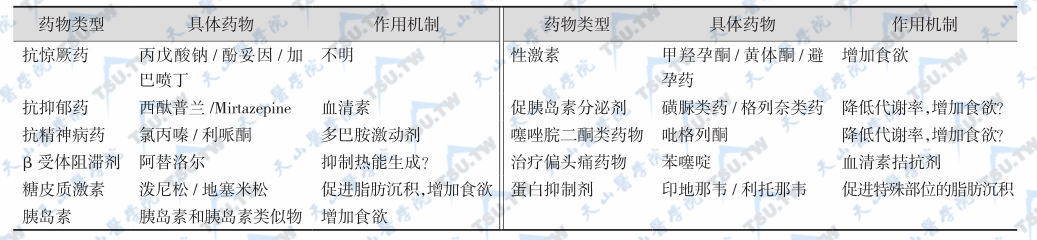
许多药物可引起肥胖,见下表。
致肥胖药物及其作用机制