
按突变酶系分类糖原贮积症
GSD因为糖原合成和分解过程中所需的酶基因突变,其表达的相应酶活性完全丧失或明显减低,引起糖原贮备减少或糖原在细胞中堆积而致病。酶基因突变包括点突变、缺失、插入和剪接突变,其中以点突变最常见。
糖原合酶突变(0型)
小鼠缺乏转录因子CCAAT增强子结合α(C/EBPα)基因则不能像正常小鼠一样贮积糖原。但用多态性微卫星侧面标志分析,排除了C/EBPα基因与GSD 0型连锁。将患者肝脏匀浆与健康人肝脏匀浆混合不能使糖原合酶激活,提示0型GSD缺陷在糖原合酶。糖原合酶基因命名为GYS2。定位于染色体12p12.2,共有16个外显子。
葡萄糖-6-磷酸酶突变
葡萄糖-6-磷酸酶(glucose-6-phosphatase,G6Pase)系统含有几种亚基。G6Pase的作用是使葡萄糖变为6-磷酸葡萄糖,由其中的催化亚基完成。G6Pase位于内质网中,由高度糖基化的催化亚基和46kD的G6P转移酶组成,均为高度亲水性蛋白,分别有9次和10次穿膜伸展区,两者相互作用而发挥生理作用,活性位点面向内质网腔。除G6P转运蛋白外,还有两个转运蛋白移位酶(translocase)T2和T3。T1的作用是将G6P转运入内质网腔中;T2将G6P或焦磷酸水解所产生的磷酸运出内质网;T3是微粒体葡萄糖转运蛋白,其作用是把G6P水解所产生的葡萄糖运出内质网。
G6P酶基因定位于染色体长臂17q21,长12.5kb,有5个外显子。酶蛋白在内质网膜中有6个伸展节段。G6P酶的特性为:①亲水性。②在微粒体中酶活性呈潜伏性,只在微粒体裂解后G6P酶活性才表达。③将微粒体制备物与肝G6P酶在pH 5.0、37℃下温育10分钟,G6P酶的磷酸水解酶(phosphohydrolase)活性即完全被灭活。④将人G6P酶cDNA转染给COS-1细胞所表达的G6P酶与人肝细胞微粒体中的G6P酶无任何区别。G6P移位酶(G6PT)T2的结构尚不清楚,G6PT基因定位于11q23,该基因长约5.3kb,含9个外显子,在脑组织和肝脏中存在两种不同的转录本,该基因突变导致Ⅰb型GSD和Ⅰc型GSD,已知导致Ⅰb GSD的突变类型有:R28H、W118AR、G339C、G339AD、E355t、R415t、(4-bpdel,2-bpINS,NT1094)、(170-bpdel,NT148)、(2-bpdel,1211CT)、(12-bpINS,NT1103)、V235delT、(IVS7,G-T,+1)、(IVS1,G-A,+1)和794G-A,其中(170-bpdel,NT148)、(2-bpdel,1211CT)和(12-bpINS,NT1103)3种在Ⅰc患者中也存在;已知导致Ic型GSD的突变类型有:(IVS8,4-bpdeL)和W96ter两种。
糖原脱支酶基因突变
糖原脱支酶(glycogen debranching enzyme,GDE)是糖原溶解时先从最外周的分支通过脱支酶作用而释放出葡萄糖。在糖原中葡萄糖的相互连接有两种形式:一种是1,4连接,即葡萄糖分子中的第1位碳原子与另1葡萄糖分子中的第4位碳原子在α位连接在一起;另一种形式是1,6连接,由第1位碳原子和第6位碳原子之间连接。这两种连接分别由糖原合成酶和分支酶(branching enzyme)催化。GDE是在1个多肽链上有两个独立的催化活性,这两个催化活性分别由寡-1,4→1,4葡聚糖(glucan)转移酶(transferase)和淀粉-1,6葡萄糖苷酶(α-glucosidase)来完成。它们之间互不依赖,独立发挥催化作用。糖原经磷酸化酶作用后,在糖原的外周支链磷酸化酶耗竭时,在支点的远端保留着4个葡萄糖基(glucosyl)残基,转移酶活性把3个葡萄糖残基从1个短的外支转移到另1个葡萄糖残基的终端,由此而使1,6连接暴露出来,然后葡萄糖苷酶使遗留的支点(α1,6连接)水解,让磷酸化酶接近α1,4连接。整个的GDE活性需要转移酶和葡萄糖苷酶两者活性均保持正常。
GDE基因称GAL基因,定位于染色体1p21。GDE的RNA由596bp的编码区和2371bp3′非翻译区组成。GDE基因有35个外显子,DNA至少有85kb。在肌肉和肝脏中的GDE酶由1个基因编码,其在肝和肌肉中的表达则由不同的遗传物质控制。在肝和肌肉中GDE mRNA通过单个脱支酶基因的RNA不同转录而产生,除了在5′端非翻译区序列不同外,其余均相同。用猪肌肉中纯化的分支酶制备的多克隆抗体作免疫印迹分析,猪和人各种组织中的脱支酶常都是160kD,但也有报告为174kD者。
磷酸化酶激酶β亚基和α亚基突变
糖原溶解需要糖原磷酸化酶(phosphorylase),而磷酸化酶则需要糖原磷酸化酶激酶(PHK)激活。编码糖原磷酸化酶激酶的基因为PHK,PHK有3种同工酶,分别由3个不同的基因编码,它们的基因座定位为PHKA2在Xp22.1p22.2、PHKB在16q12-q13 和PHKG2在16p11.2-p21.1。肝和肌肉中的糖原磷酸化酶均由α、β、γ和δ4个亚基组成。此酶活性受α和β亚基中的特异性残基的磷酸化的调节,钙则通过δ亚基来调节酶的活性,γ亚基则具有催化活性。
其他类型
酸性α-糖苷酶缺陷导致Ⅱ型GSD(pompe病),糖原分支酶缺陷导致GSDⅣ型(Anderson病),肌肉磷酸化酶缺陷导致Ⅴ型GSD(McArdle病),肝脏磷酸化酶缺陷导致Ⅵ型,GSD磷酸果糖激酶缺陷导致Ⅶ型GSD(Tarui病),磷酸甘油转换酶缺陷导致Ⅷ型GSD。
糖原贮积症分为十型和若干亚型
由于不同的酶先天性缺陷,故根据酶的缺陷可将本病分为许多类型,见下表。
糖原贮积症的分型
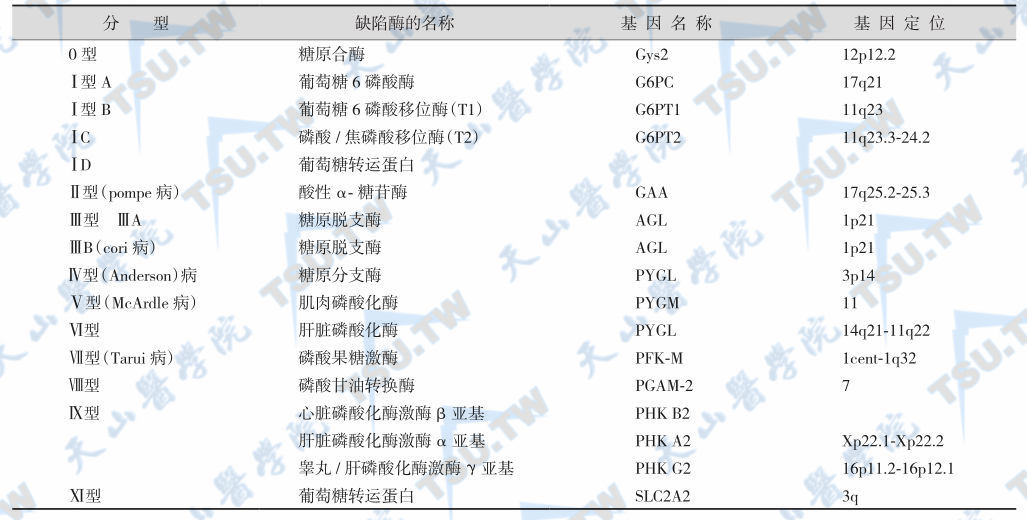
以Ⅰ型GSD最常见。有些类型的GSD由于缺陷酶由多种亚基或异构酶组成,因此可有多种亚型,Ⅺ型病例最少。
