
英文:Turner Syndrome;
同义名:女性型性腺发育不全,X单体综合征,先天性卵巢发育不全综合征。
溯源:1938年Turner首先描述过本病的主要临床症状。1954年Polani发现本综合征患者的间期细胞核中,不像正常女性那样有二个X染色质(Barr小体)。1959年Ford确证本病患者虽然外表呈女性,但缺少了一条X染色体,其核型为45,X。故又称X单体综合征。
发病机制
主要病因是女性患者的性染色体不是两条,而是缺少一条X染色体(45,X),或x染色体之一的部分片段发生缺失,如Xp-、Xq-、i(Xq)、i(Xp)等。绝大多数源自新发生的染色体畸变。新生女婴的发病率为1/2500,但在自然流产儿中的发生率为7.5%,表明45,X的胚胎多在胎儿期死亡或流产。
- 遗传学:本病的最常见的核型为45,X(占55%以上),约25%为X染色体的各种结构重排,如46,X, i(Xq); 46, X, i(Xp); 46, X, del (Xp); 46,X,del(Xq); 46,X,r(X)等。约10%为45,X/46,XX;45,X/47,XXX等。在X染色体上已明确定位的基因有100多个,其中与性别发育异常有关的基因主要有XY女性型性腺发育不全基因(p21→p22),原发性性腺发育不全基因(p21.2→p21.3)和睾丸女性化基因(p11→q11);Aarskog-Scott综合征(面部、生殖系统发育不全,基因定位于q13)等。
- 皮纹学:三叉点扁位(t'),指纹中斗形纹比例高,故TFRC亦高,一般≥200。
临床表现
表型女性,身材矮小,成人身高一般不超过150cm。卵巢萎缩呈索条状,原发性闭经,不育。乳距宽,乳房不发育,盾状胸。子宫小,外生殖器发育不良,成年后仍呈幼稚状态,阴毛、腋毛缺如或稀少。蹼颈,后发际低,肘外翻,新生儿期手、脚常呈淋巴水肿。第4、5指(趾)骨与掌(跖)骨短或畸形。面部、背部等有多发性黑痣。患者一般智力正常,少数智力稍差。
嵌合型病例的临床症状一般较轻,其中20%左右可有月经。X染色体有结构异常的病例中,具有X染色体短臂等臂即46,x,i(Xp)或x染色体长臂部分缺失即46,X,Xq-的病例(两者均有X染色体长臂缺失或部分缺失),一般身高正常,有月经,只有30%的病例具有本综合征的各种典型症状;而46,X,i(Xq)或46,X,Xp-的病例(两者均有xp的缺失或部分缺失),则身体矮小和具有性腺发育不全的各种症状。根据近期报道的10例X染色体短臂缺失的病例的分析表明,如果是Xp21→pter缺失,一般可以生育,但如果是Xp11→pter缺失则不孕,据此可以推测,Xp11片段对卵巢的发育和功能具有重要作用。
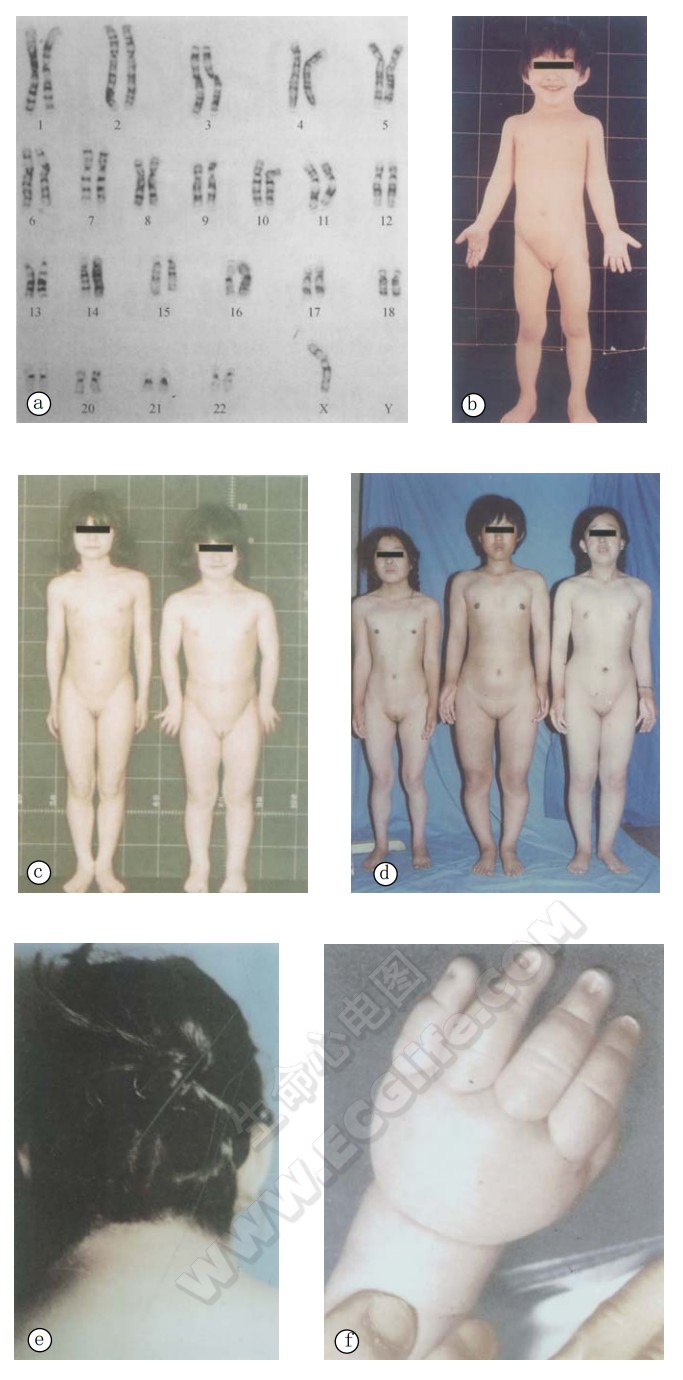
- a:患者核型(45,X);
- b:示8岁女孩;
- c:姐(左,9岁)、妹(右,7岁)患者;
- d:19岁(左)、24岁(中)、21岁(右)患者外观:体矮,蹼颈,肘外翻。青春期后外生殖器仍呈幼稚型,乳距宽,乳房不发育,无阴毛,腋毛,主动脉狭窄(b、c一姐姐);
- e.后发际低(b);
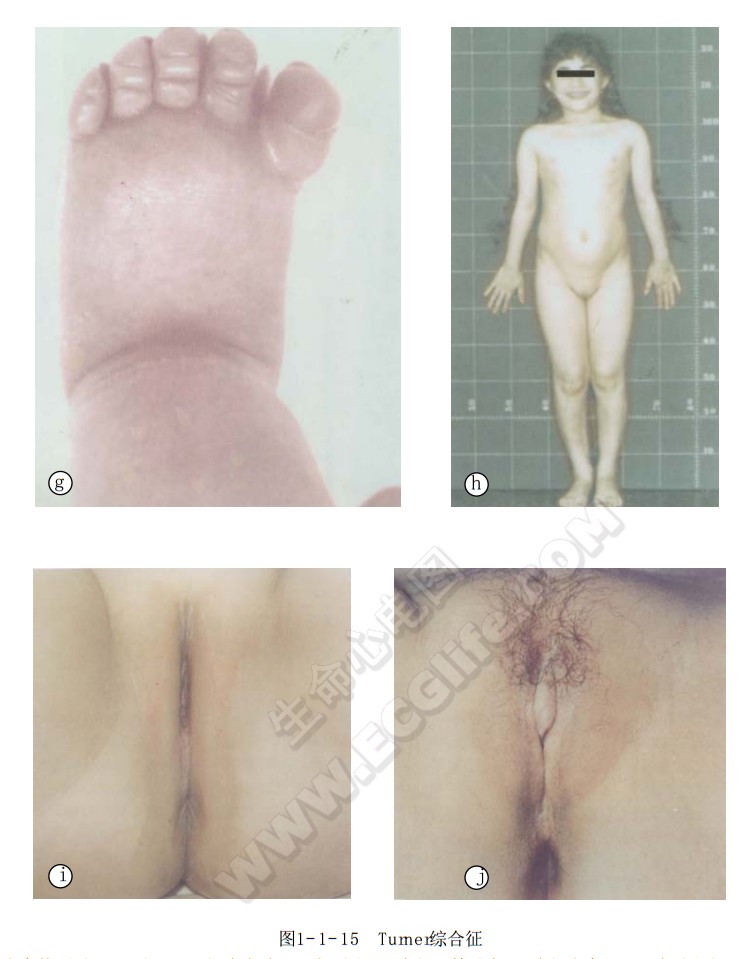
- f、g:示2月龄患儿的手、脚部呈淋巴水肿;
- h:21岁患者(d图右);
- i:示d图中右侧患者未采用雌性激素治疗前的外阴;
- j:示采用雌性激素治疗4个疗程后,外阴性征明显改善,身高亦增长了6cm。
心血管损害:约30以上病例有先天性心脏病,主要为主动脉狭窄(占50%以上);其次为主动脉双瓣叶、主动脉下狭窄、室间隔缺损、二尖瓣脱垂等;约5%~10%伴发与主动脉狭窄无关的高血压。近年来Miller等应用超声心动图技术发现,本综合征患者中34%伴发无狭窄的二叶主动脉瓣。
诊断
主要依据:
- 核型分析,X染色质阴性;
- 体矮,蹼颈,肘外翻,后发际低;
- 卵巢、子宫发育不全,外生殖器幼稚,原发性闭经,乳距宽,盾状胸,乳房不发育,无阴毛、腋毛;
- 新生女婴手背、脚背有明显淋巴水肿,皮肤有黑痣;
- 心脏畸形。
预后
伴发心脏病的患者多在婴儿期死亡。其余多数可存活。对存活者应及时给予激素替代疗法治疗。例如在妇科医师指导下,从16岁左右开始采用雌性激素治疗,可以改善一些临床症状,如身高增加,促进第二性征如乳腺、外阴、阴毛的发育等。
