
英语:partial 18q trisomy syndrome;
溯源:1972年由Rott等首次报道,1977年由Turleau等确认为18q部分三体综合征。
发病机制
病因是第18对染色体之.的长臂部分片段重复。其中q21片段的重复对本症的临床症状有重要作用。临床特征与18三体综合征类似,但症状较轻。
- 遗传学:大部分病例源自亲代的染色体平衡易位携带者,少数为新发生的染色体畸变。已报道的核型有:
- 46,XX,der(6),t(6;18)(p11;q22q23);
- 46,XX,der(6),t(6;18)(p25;q21)pat;
- 46,XX,der(13),t(13;18)(p13;q12)mat(pat);
- 46,XX,der(21),t(18;21)(p11;q11)mat;
- 46,XY,dup(18)(pter→q12::q12→qter)等。
- 皮纹学:通贯手,10指均为弓形纹,故TFRC为O。
临床表现
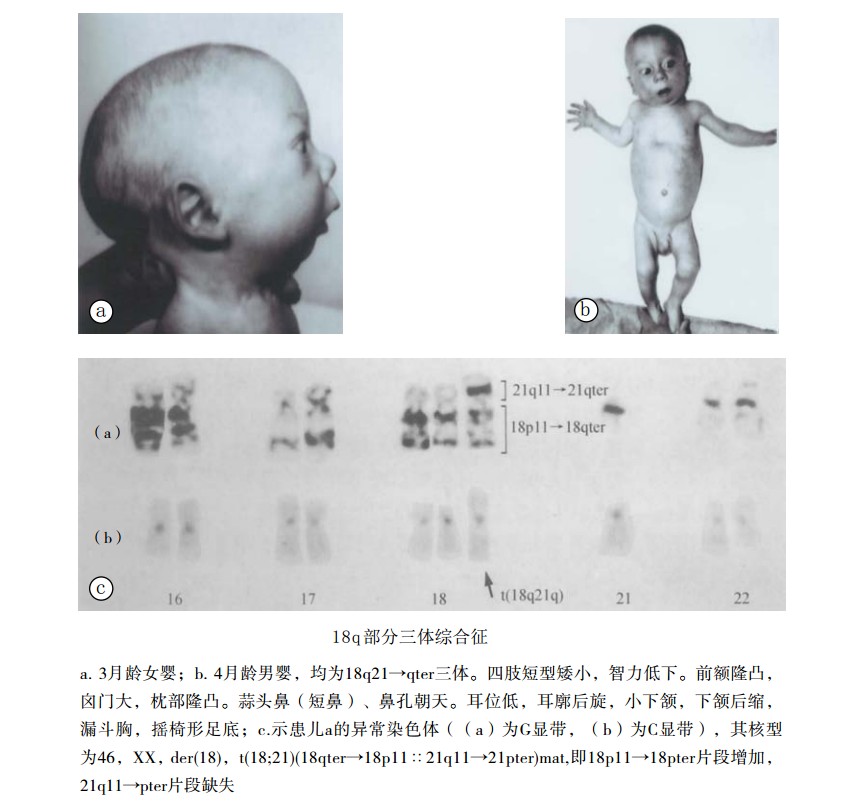
出生前后生长发育迟缓,身材矮小,智力低下。前额隆凸,枕部隆凸。蒜头鼻(短鼻),鼻孔朝天。耳低位、后旋。腭弓高,小下颌,下颌后缩。蹼颈,肾畸形,摇椅形足底。
心血管损害:可有先天性心脏病,如房间隔缺损等。
诊断
主要依据:
- 核型分析;
- 基本特征与18三体综合征相似,但本病伴发的畸形少,而且有蒜头鼻(短鼻),10指均为弓形纹和一般婴儿期仍存活的特点,但是对两者进行鉴别时,主要应根据核型分析。
预后
预后比18三体综合征好,存活者也比18三体综合征多,已有数例成年病例的报道,应加强婚、育的优生指导。
系统的医学参考与学习网站:天山医学院, 引用注明出处:https://www.tsu.tw/heart/zhz/rst/yichang/620.html
