
英文:Bloch-Sulzberger syndrome(MIM 308300);
同义名:色素失调症Ⅰ型,色素失禁症,真皮变色性色素沉着症,Bloch-Siemens综合征,色素颗粒细胞痣。
溯源与发展
1906年由Garrod首次描述了本病,继之于1925年Bardach以序列痣为题目报道了2例。其后1926年Vaegeli和1929年Siemens还分别以色素噬细胞痣和真皮变性黑变病为名称报道本病。但多数学者认为1926年Bloch和1927年Sulzberger分别报道同一2岁女孩患有先天性色素异常,病变多分布于臂部、踝部皮肤,色素斑酷似人工画笔描绘而成,而采用色素失调症(incontinentia pigmenti)命名。我国于1956年由朱德生报道2例之后,陆续有个例报道。其特征为皮肤病损、神经系统、眼部症状、齿及骨骼系统异常。
发病机制
一种皮肤色素紊乱伴有眼睛、牙齿,骨骼和心脏畸形的综合征。1961年Lenz根据病例分析提出本症是一种男性致死的X连锁显性遗传病。1977年Kunze等在一个47岁,XXY (Klinefelter综合征)的男性病例中具有色素失调症症状,亦与Lenz的假设相符。1987年Ormorod等描述的一个47岁,XXY病例的家系,其家系系谱提示其遗传方式显示是男性致死的X连锁的显性遗传,此家系中受累女性的表现型与Lyon的假设的X染色体随机失活相符。因为在46岁,XX的女性患者中,女性的两条X染色体中,一条是正常的;一条是带有致病基因的(称杂合子),故虽然有色素失调症症状,但对生命无严重影响,而46岁,XY的男性患者,只有一条X染色体(称半合子),当这条X染色体上带有致病基因时即严重发病,多死于官内,故临床上男性患者少见。
- 遗传学:XD遗传方式,群体中男女发病率约1:37。致病基因定位于X染色体的Xp11.21,Ⅱ型Xq28。
临床表现
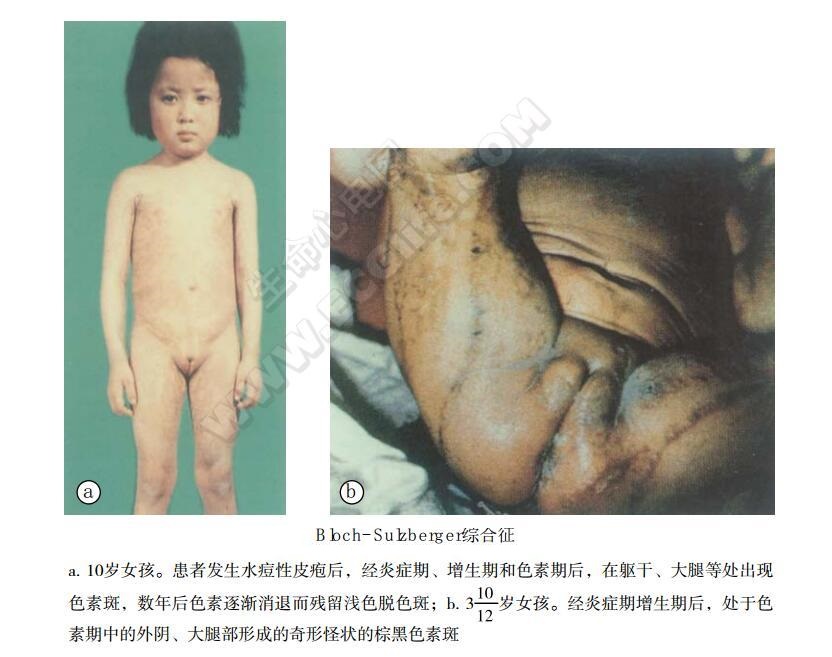
临床所见病例多为女性。多数在生病第1周起病,迟至6龄发病的很少,偶见于成年人。先于躯干两侧沿神经分布出现荨麻疹或水疱性皮疹,经炎症期,增生期和色素期后,在躯干,大腿两侧出现奇形怪状的棕黑色色素斑,可反复发作。色素斑可持续数年,色素斑消退后残留浅的脱色斑。血中嗜酸性粒细胞增高(5%~56%),水痘中酸性粒细胞可高达90%以上。小头,智力低下,可伴发癫痫,四肢瘫痪。小眼球、白内障,视神经萎缩失明。1岁后的患者2/3有牙齿异常(部分无牙或完全无牙),指(趾)甲萎缩。另外可有出牙延迟,圆锥状牙冠、缺牙,头部毛发异常、脱发、毛发短,指(趾)甲缺损及骨骼异常,如脊柱裂、多余肋、髋关节脱臼等。
- 心血管损害:先天性心脏畸形,多为动脉导管未闭,原发性肺动脉高压,心脏功能不全,继发性肺心病。
辅助检查
水疱内嗜酸性粒细胞占95%,外周血嗜酸性粒细胞占74.5%。患者体内血中可测到胞浆抗体。
诊断、鉴别
- 诊断:依据典型炎症期、增生期、色素期三阶段临床特征以及出生不久发病,大部分为女性,同时伴有神经系统、眼及骨骼改变,心血管损害;结合病理及实验室检查可以确诊。
- 鉴别:需与神经纤维瘤、色素性麻疹、大疱性表皮松懈症、儿童期大泡性病、天疱疮相鉴别。本病尚需与Franceschi-Jadasson综合征鉴别。后者特点:
- 色素沉着为网状;
- 无前区炎症变化,无水泡及疣状损害;
- 男性发病率高;
- 为显性遗传。
治疗及预后
无特殊疗治。通常色素失调症终末阶段色素于2岁开始消退,到成年几乎认不出。皮肤病变可自然消退不必治疗。水泡期应防止继发感染,可试用肾上腺皮质素类。若合并癫痫进行癫痫治疗。女性患者多为良性过程,到青春期可能痊愈。
