
本病是中胚叶和外胚叶结构发育障碍,表现为广泛的进行性皮肤和骨骼缺陷。1920年Jessner首次报道本病,1962年Goltz报道3例,并复习文献,正式命名此病,故又名戈尔茨综合征(Goltz syndrome)。
本病几乎累及女性,提示为X连锁的显性遗传,但也有X性染色体易位及环境因素致病的报道。大多数病例是散发的。少数病例为男性患者,由于仅有一条X染色体,病情常较重而易死于子宫内。有学者发现FDH基因定位于9q32-qter。
临床症状
临床上主要表现为皮肤、骨骼、齿、毛、甲等组织发育缺陷。
(一)皮肤缺陷 皮肤损害常在出生时就有,由于表皮发育不良,形成境界清楚的皮肤变薄。皮肤凹陷在某些部位,脂肪可由真皮缺陷部位向外突出,形成脂肪疝,呈柔软的黄色结节,线状排列。在臀、股、腋部还可看到线状或蛇行排列的褐色色素斑,伴网状和筛状萎缩、毛细血管扩张的红斑,犹如皮肤异色病改变。线状损害常沿Blaschko线分布,最常见于肢体或颈部。有些患者在出生时某些部位的皮肤可完全缺如,像先天性皮肤再生不良。
另一特征性的皮肤表现在唇、肛门、阴道口周围有小的红色进行性发展的乳头状瘤,容易被误诊为尖锐湿疣。
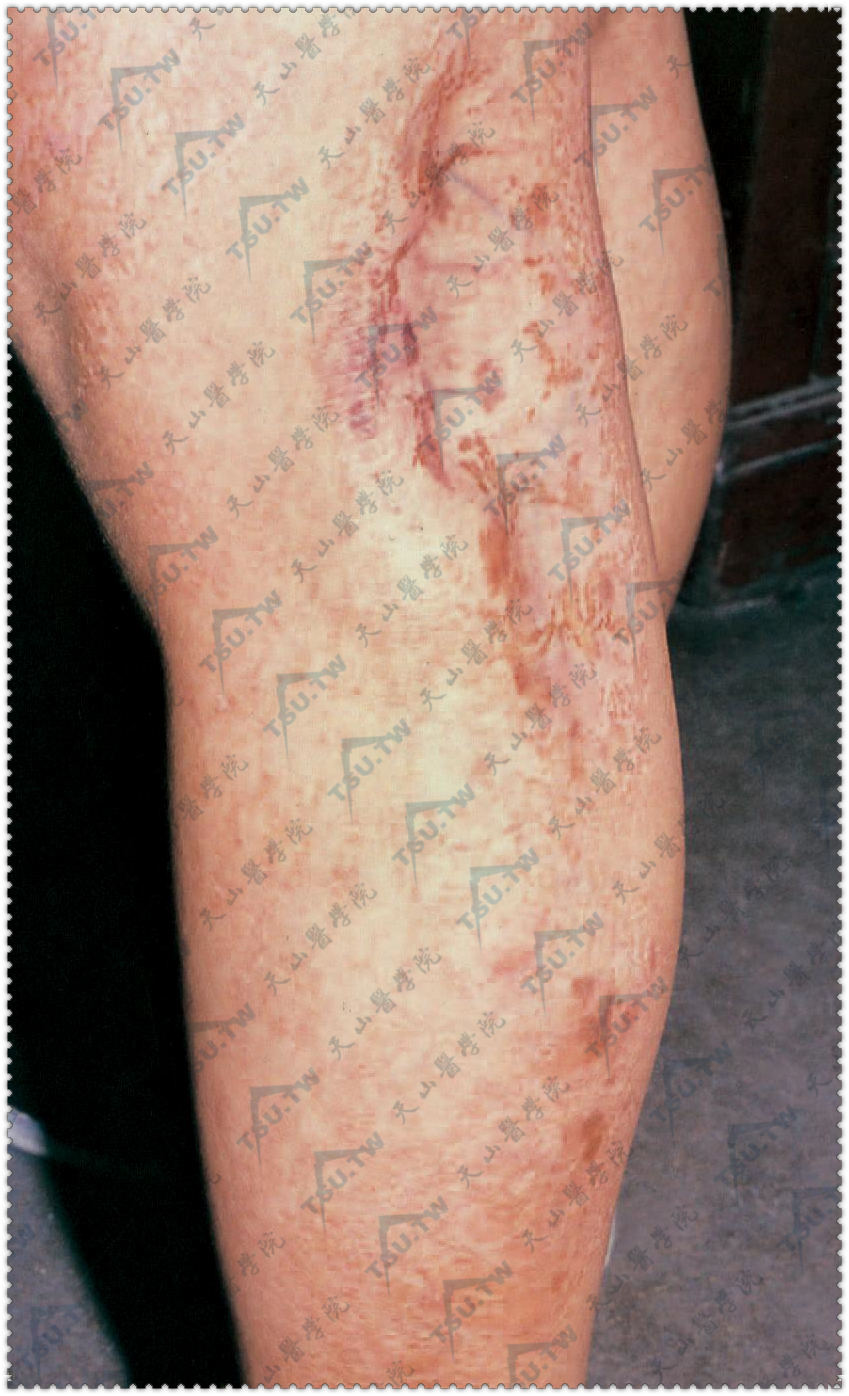
下肢局灶性皮肤变薄,境界清楚,表皮凹陷,见线状红褐色斑
其他皮肤表现还有风团、皮肤划痕症、水疱、跖部角化、嘴角周围放线状沟纹、出汗异常、光敏等。
(二)骨骼缺陷 最常见的是指(趾)骨的改变,可有无指、缺指或并指,如龙虾爪(lobster-claw),可见于60%~70%的患者。脊柱也常累及,如侧凸、后凸,腰椎骶骨化、脊柱裂。此外,有右锁骨发育不全,头颅发育不全、不对称等。一般来说,骨骼结构发育不良常伴有其上方的皮肤发育不良。X线检查有特征性的表现,在长骨干骺端呈条纹状改变及耻骨联合变宽,有诊断价值。
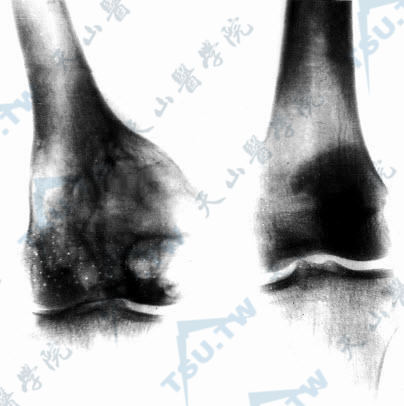
X线检查见骨骺端呈现条纹状改变
(三)牙齿缺陷 齿发育不良,表现为齿数和形态异常,如小齿、生长迟缓、牙齿变形、缺齿等,可见于40%以上的患者。
(四)毛发和甲 毛发稀少而易脆裂,在严重萎缩和发育不良区域,毛发可完全缺如。耻骨及两腋部可见斑状脱毛区,可有缺甲、甲结构异常、甲营养不良而呈匙状甲、沟状甲等。
(五)其他 眼部常受累,虹膜、睫状体、视网膜或全眼球缺陷是特征性的,偶有小眼球或一侧眼不发育,泪管异常、眼外肌病变。耳朵变形,耳软骨缺陷,可伴双侧感觉神经性及传导性耳聋。约有1/3病例身材矮小,精神发育迟缓。此外,心、肾、中枢神经系统损害亦可见到。
组织病理
表皮正常,真皮发育不全,胶原束减少、变细,弹性组织减少并被脂肪组织所代替。在损害较轻的部位,在真皮血管及附属器周围仍可见少量狭窄的纤维瘤结缔组织带,但胶原纤维细小,呈低发育状态。皮肤附属器变少。黏膜周围乳头瘤损害呈纤维血管乳头瘤的病理表现。
诊断及鉴别
根据带状排列的皮肤异色病表现及“脂肪疝”形成、骨骼畸形、X线检查有特征性骨改变,可诊断。但应鉴别:
- 色素失禁症 有特征性水波状皮肤色素改变,无脂肪疝及萎缩表现。
- 线状表皮痣伴骨骼和中枢神经系统缺陷 组织病理可鉴别,呈表皮痣改变,伴角化过度,无真皮改变。
- 先天性皮肤再生不良 呈局限性境界清楚的皮肤缺如,可自然痊愈,无其他系统发育不良表现。
- 先天性皮肤异色病 异色症表现明显,且不呈带状分布,可伴光敏、白内障、侏儒及生殖器异常。
- 浅表脂肪瘤样痣 组织病理与本病很相似,但临床表现不同,呈单一或成群密集的丘疹或斑块,真皮内有脂肪组织。
预防及治疗
外科矫形治疗。
