
本病又名斯威特综合征(Sweet syndrome)、斯威特病(Sweet's disease)。1964年Sweet首先报道了6例女性患者。本病主要表现为发热,四肢、面、颈部有疼痛性红色丘疹、斑块或结节。末梢血中性粒细胞增多,组织病理学真皮有密集的中性粒细胞浸润。
病因及发病机制
病因尚不明确。50%的病例与潜在的疾病相关。可能与以下因素有关:
- 感染:患者于发病前可有上呼吸道感染,如咽炎、扁桃体炎、支气管炎、流感综合征等。
- 药物:如粒细胞集落刺激因子(G-CSF)、全反式维A酸、米诺环素、甲氧苄啶-磺胺甲
 唑、卡马西平、肼屈嗪和口服避孕药。
唑、卡马西平、肼屈嗪和口服避孕药。 - 肿瘤:20%~25%的病例与恶性肿瘤有关。大部分为血液系统的恶性肿瘤,如急性髓源性白血病、淋巴瘤,还见于贫血或红细胞增多症等。15%左右的恶性肿瘤为实体瘤,如泌尿生殖道、乳腺(女性)和胃肠道(男性)来源的。该病的皮肤表现可以是恶性肿瘤的最初的表现或先于诊断恶性肿瘤数月到数年。如果该病反复发作可提示潜在肿瘤的复发。
- 与其他疾病伴发:如白塞病、结节性红斑、结节病、类风湿关节炎和甲状腺疾病。可能是两病同时存在,而不是真正的疾病的相关性。应与类风湿嗜中性皮病的表现相区别。
- 亦有发生于皮肤外伤后。
该病的致病机制不明。可能机制是对细菌、病毒、药物或肿瘤抗原等的超敏反应。有以下几个发病机制参与:①免疫复合物即Ⅲ型变态反应。给本病患者皮内注射细菌抗原(草绿色链球菌)和真菌抗原(白念珠菌)后,复制出的改变在临床与组织病理变化均与原损害相同,提示细菌或真菌抗原与相应抗体形成免疫复合物,引起血管及其周围炎症,故可能是一种局部Arthus反应。研究发现血管壁内有免疫球蛋白及补体的沉积,但结果也不一致,靳培英检查20例,发现16例有免疫球蛋白(IgG 4例、IgM 7例)和(或)C3沉积,14例沉积于血管壁。循环免疫复合物增高,10例超出正常范围。认为本病是白细胞碎裂性血管炎的一个轻型或亚型。但也有认为血管炎的发生是继发性的。②T细胞激活作用的改变。有Th细胞分泌的细胞因子的失衡,认为Th1细胞因子(IL-2和INF-γ)可刺激细胞因子的级联反应,导致中性粒细胞的激活,并释放出毒性代谢产物。③中性粒细胞功能的改变。④部分病例有ANCA存在。
临床症状
该病多见于中年以上女性(40~70岁),男女之比是1:3,但50岁以上人群中,男女发病率相等。也可发生于婴儿,甚至新生儿。夏季好发,欧洲病例在春秋季好发。依照其发病机制,临床上可分为四类:
- 经典型或特发型(71%);
- 恶性肿瘤相关型(11%);
- 炎症性疾病相关型(16%);
- 妊娠相关型(2%)。四个亚型临床表现很相似。
经典型或特发型 常发生于中年女性,与感染、溃疡性结肠炎等有关。特征性临床表现有高热、中性粒细胞增高和血沉加快,皮肤表现包括迅速发展的境界清楚的红色至紫色疼痛性结节、斑块。结节逐渐扩大、增多,颜色变深,疼痛加重;斑块扁平隆起,直径2~10cm,边界清楚而陡峭,表面可因真皮乳头高度水肿呈乳头状或粗颗粒状,似假性水疱。部分斑块表面可见散在因为中性粒细胞移入表皮而形成的针尖大小的水疱或脓疱,表面有结痂,但不发生糜烂或溃疡,触之较硬,有触痛(下图)。有的斑块中央部分渐渐消退而有鳞屑与色素沉着,周围可远心性扩大而呈环状损害,有的斑块中央变为淡黄色,而呈靶样损害。皮损好发于面、颈、躯干上部和四肢。可不对称分布,单发或多发,可局限于某个部位,也可泛发。后者可见于恶性肿瘤相关者。皮损经1~2个月自行消退,局部不留瘢痕,仅有暂时性褐色色素沉着。本病常易复发,有潜在恶性病的更易复发。
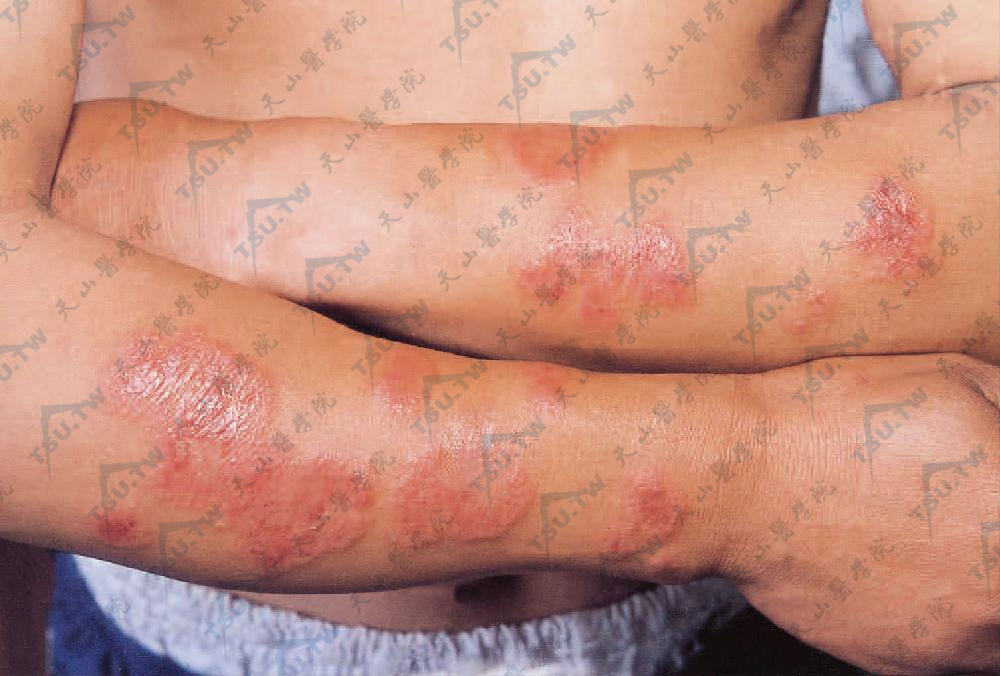
两前臂多个圆形或类圆形红色斑块,边缘可见粟粒至绿豆大小假性水疱
3/4以上的病例有系统症状。85%~90%病例伴有发热和不适,尤其是伴有恶性肿瘤的患者,部分患者可有关节痛、关节炎,约占25%~50%,或可有肌痛,另有32%~75%的患者有眼结合膜炎、浅表性巩膜炎。类似于阿弗他溃疡的黏膜损害的发生率在经典型为2%或3%,而伴血液系统恶性疾病的病例为10%。肾脏受累者占11%~72%,表现为蛋白尿(11%~16%)、血尿、颗粒管型及肌酐清除率异常,肾活检见局灶性肾小球系膜细胞增生,局灶性基膜增厚及间质圆形细胞浸润。少数还可有神经、肺、肠道、肝脏的累及。还可发生多灶性无菌性骨髓炎。
妊娠相关型 常发生于妊娠的前3~6个月。皮损常发生于头、颈和躯干,很少发生于上肢,下肢可发生结节性红斑样的损害。可自行消退。在下次妊娠时又可以再发。
药物相关型 最常见的是粒细胞集落刺激因子,本病可认为是该药在皮肤的药理学并发症;用于治疗前髓细胞性白血病的全反式维A酸,约使用2周后即可发生本病。
恶性肿瘤相关型 常无中性粒细胞升高,且常复发。本病常伴有末梢血白细胞计数增高(60%),70%病例有中性粒细胞比例增多,白细胞左移(50%),或白细胞总数不高而中性粒细胞比例增多,在与恶性肿瘤伴发的病例,大部分有贫血(男女分别为93%和71%),半数病例有血小板减少。血沉常增快(90%)。免疫球蛋白与补体测定多为正常。针刺反应阳性率达80%。少数病例可有ANCA阳性,但ANCA不是该病的血清学标志。
组织病理
表皮常正常,可有轻度角化不全,海绵形成,极少数病例可有中性粒细胞移入到表皮,形成角层下脓疱(20%)。主要变化在真皮,真皮乳头明显水肿,偶可形成表皮下疱,真皮浅、中层毛细血管扩张,内皮细胞肿胀,真皮上部密集的以中性粒细胞为主的浸润,有中性粒细胞核固缩和碎裂(核尘),间有淋巴细胞、嗜酸性粒细胞和组织细胞,也有报道开始即为淋巴细胞浸润。浸润常为弥漫性,真皮全层甚至皮下均可有类似浸润。也可在血管周围、汗腺周围或真皮上部呈带状分布。陈旧皮损真皮中性粒细胞浸润明显减少,而淋巴细胞和组织细胞相对增多。
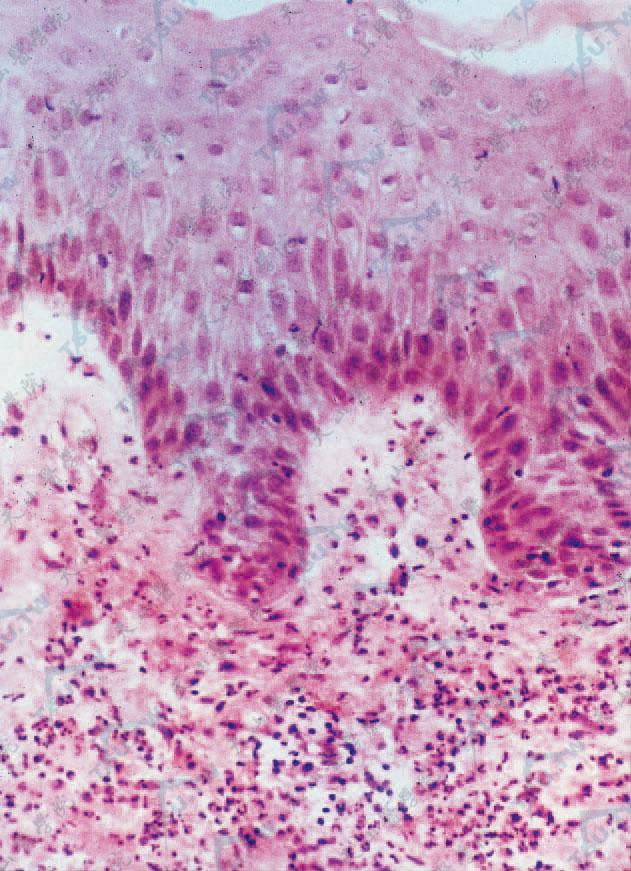
真皮乳头显著水肿,真皮内白细胞弥漫性浸润伴有核碎裂(HE染色×400)
诊断及鉴别
根据本病特有的临床表现,结合组织病理变化,诊断并不困难。本病患者可以不发热,血中白细胞计数及中性粒细胞亦可不增多,血沉也可正常,这些表现均不作为诊断的必要条件。本病的诊断标准见下表。
急性发热性嗜中性皮病诊断标准(Su&Liu修订版,1986年)

本病需与持久性隆起红斑、变应性皮肤血管炎、多形红斑相鉴别(下表)。还应与白塞病、肠吻合综合征、结节性红斑等鉴别。白塞病有口腔、生殖器溃疡,眼部症状如眼色素层炎,可有毛囊炎样或多形红斑样损害;肠吻合综合征主要有脓疱性损害,病理表现为嗜中性皮病改变,此前有胃肠手术或胃肠道疾病的病史对鉴别很重要;结节性红斑多发生于中青年女性,主要表现为双小腿伸侧为主的疼痛性红色结节,表面不发生假性水疱,组织病理学表现为脂肪小叶间隔性脂膜炎改变;坏疽性脓皮病,开始为丘-脓疱疹,然后发生溃疡,而本病不发生溃疡。
急性发热性嗜中性皮病鉴别诊断表

治疗
通常用抗生素治疗效果不好。系统用糖皮质激素疗效好。泼尼松开始用量每日30mg(40~60mg/d),几天内发热及皮损即可消退。以后渐减量至停药。一般疗程需4~6周,但有时需用低剂量长期维持,以防止复发。碘化钾、秋水仙碱及雷公藤制剂亦可获满意效果,可作为轻型的一线治疗。吲哚美辛和氯苯酚嗪疗效不如碘化钾和秋水仙碱。氨苯砜、多西环素和环孢素也有效,但治疗期间需注意不良反应的发生。还有报道用依曲替酯或IFN-α治疗有效的。也可外用或皮损内注射糖皮质激素或作为辅助疗法治疗局限性皮损。
