
病因及发病机制
色素失禁症(Incontinentia Pigmenti)也称为Bloch-Sulzberger病,主要见于女性,是一种X连锁显性遗传性疾病。目前证实为定位于X染色体长臂的Xqll(IP1)和Xq28(IP2)突变引起。核因子(NF)-κB基因调节体(NEMO)基因突变在抑制肿瘤坏死因子诱导的细胞凋亡中起作用,显示其是发生本病的原因。NEMO基因突变的老鼠模型已经建立,其可以导致雄性死亡而杂合子雌性伴有类似色素失禁症的皮损。患病动物的皮损活检显示角质形成细胞凋亡增加、炎症和色素失禁。在本病患者受累皮肤的基底层上皮内,嗜酸性粒细胞趋化因子(一种NF-κB活化的趋化因子)呈强表达。这种表达伴有表皮上部嗜酸性粒细胞聚集,提示这种趋化因子具有致病作用。
临床症状
色素失禁症是一种罕见的系统性疾病,女性发病倾向显著,因为异常基因位于性染色体上,女性因存在于另一个X染色体的正常基因可将其掩盖,故症状表现不甚严重;而男性仅有一个X染色体,因而病变表现严重,多半于胎儿期即死亡。
患者出生后1周左右,于躯干两侧发生荨麻疹样、水疱样、疣状皮炎病变。继发色素性斑疹,常好发于躯干、上臂及大腿。色素沉着有如撒胡椒面或喷泉状,损害不沿皮纹或神经分布。色素可持续数年,消退后不留痕迹,或稍留淡的色素脱失斑。
临床分为三期:第一期有红斑及大疱,排列成行,出生时即有或出生后2周内显著,常波及四肢和躯干,不侵犯面部。第二期由角化过度性的疣状丘疹和斑块组成的损害,见于2/3患者,是继水疱后在相同部位出现的损害。疣状损害可类似线状表皮痣。尽管这些损害通常在1岁时消失,但可持续多年。有广泛播散、不规则散布或如涡轮状的色素沉着。第三期损害表现为奇特的网状色素沉着,以躯干部损害最显著。典型者乳头处色素沉着过度,腹股沟和腋部受累最有特征性。其后几年,损害可逐渐减轻乃至完全消退,至成年期通常不易察觉。
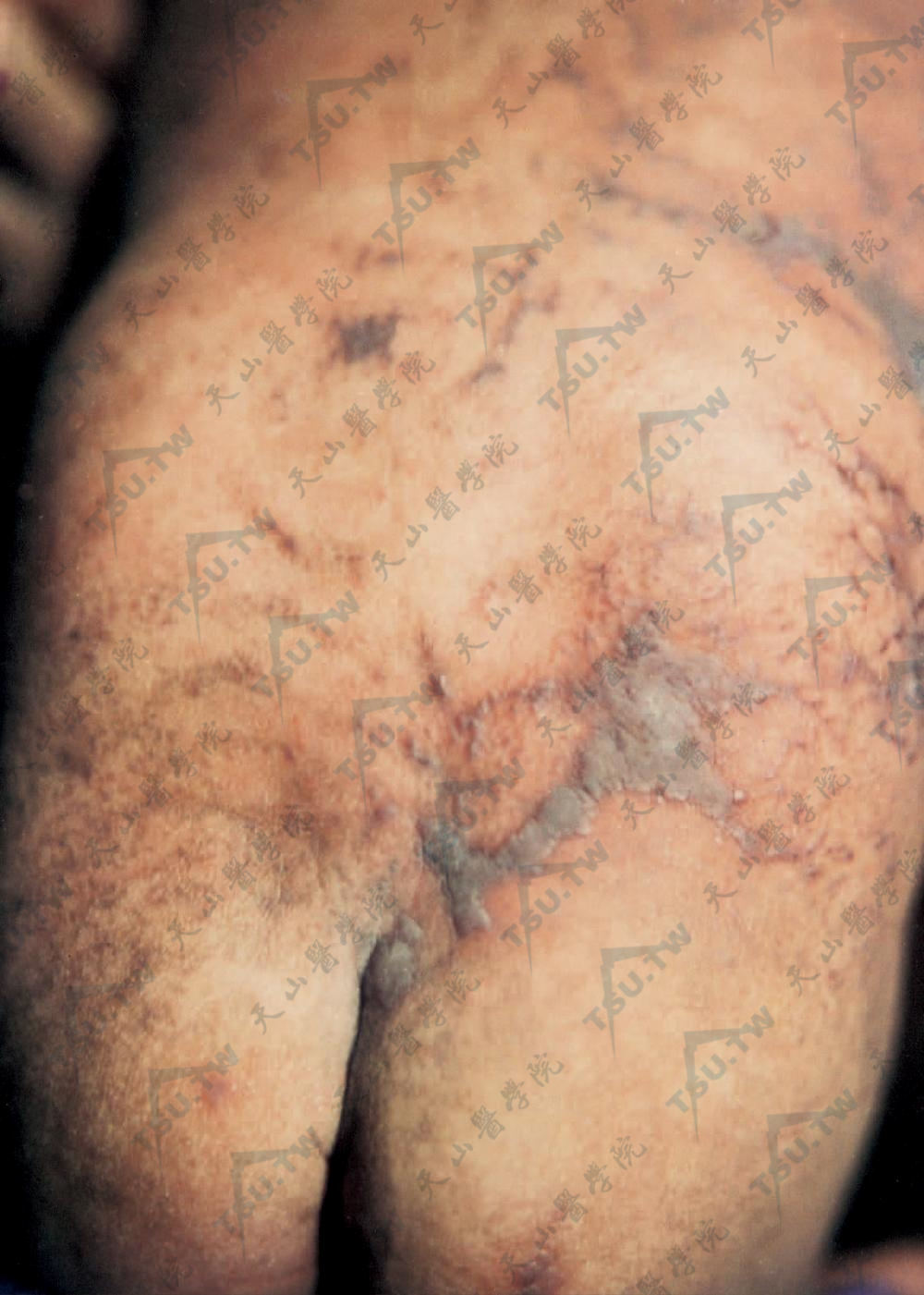
背部见红斑、水疱及苔藓样丘疹,呈条索状、流水状,皮损中见色素沉着
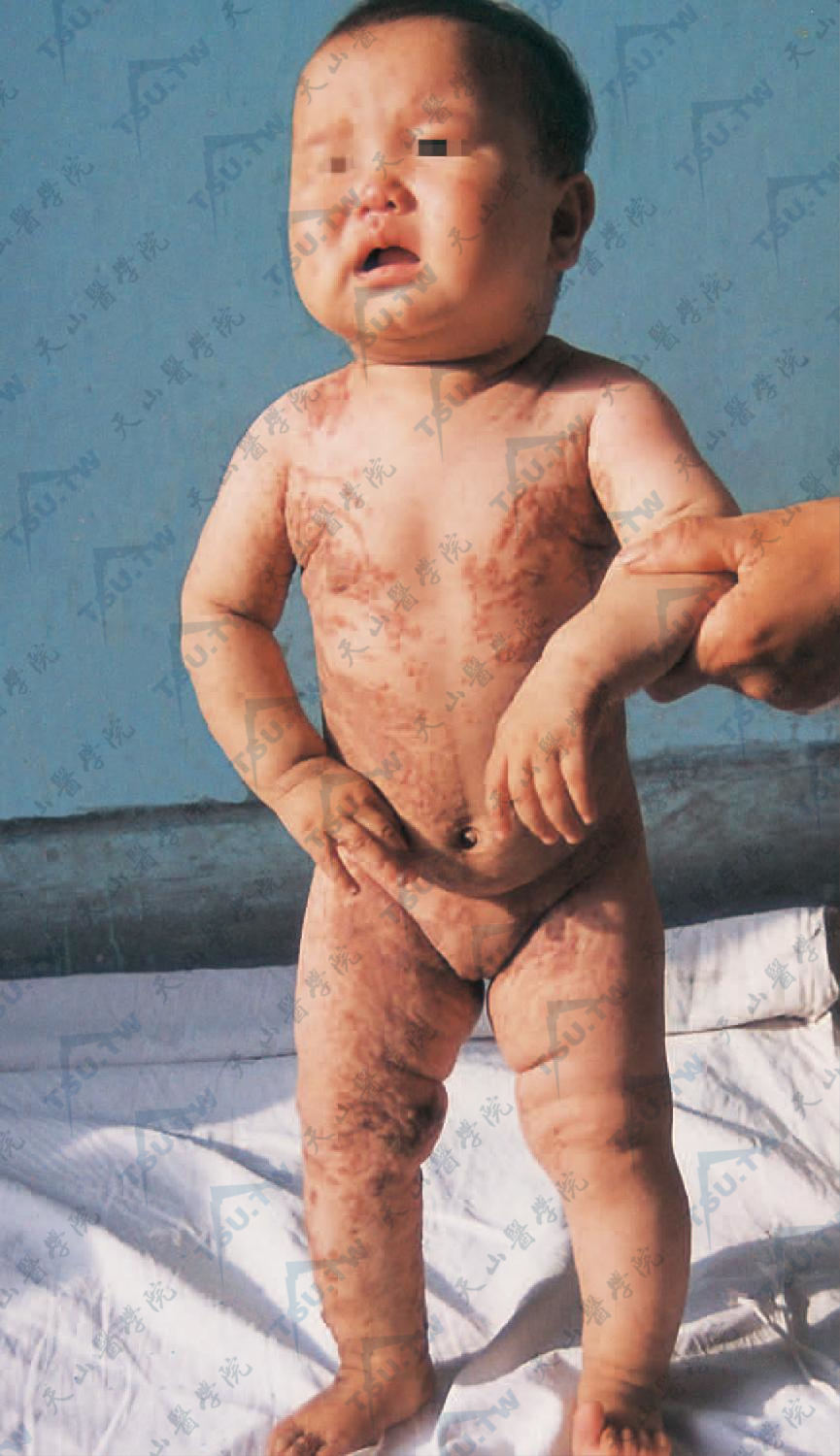
上图:躯干、上臂及大腿见荨麻疹样、疣状炎症色素性斑疹,稍隆起。皮损分布形状特殊,呈线状、流水状、喷泉状
其他皮肤改变包括假性秃发、慢性萎缩性肢端皮炎样的皮肤萎缩、甲萎缩、甲营养不良、甲下肿瘤伴其下的溶骨性损害及掌跖多汗。70%~80%的患者有皮肤外表现,多累及牙齿、中枢神经系统、眼睛和骨骼。牙齿异常可表现为出牙迟、牙变尖或畸形、脱落等。中枢神经系统的异常表现为癫痫、智力迟钝、小头畸形、脑病和运动迟缓。眼部损害中有半数较为严重,包括斜视(最常见)、白内障、视网膜脱离、视神经萎缩、渗出性脉络膜视网膜炎等。骨骼异常包括并指、颅骨变形、侏儒症、脊柱裂、畸形足、多肋骨、偏侧萎缩及腿臂缩短。
已有报道,中性粒细胞趋化性缺陷和IgE升高的免疫功能障碍,水疱期可有明显的血嗜酸性粒细胞升高。
组织病理
- 第一期:水疱位于表皮内,有海绵形成,属皮炎型水疱,疱内或疱周表皮中具有许多嗜酸性粒细胞,水疱间的表皮内侧常有呈涡轮状排列的表皮细胞和散在的大的嗜酸性角化不良细胞。真皮内有炎症浸润,有多数嗜酸性粒细胞和单一核细胞浸润。
- 第二期:表皮棘层肥厚,呈不规则乳头瘤样增生,角化过度,常有表皮内角珠,并有呈涡轮状排列的角质形成细胞和散在的角化不良细胞。真皮内轻度慢性炎症浸润,常含有少数载黑素细胞。
- 第三期:色素沉着区内真皮上部的载黑素细胞内有广泛的黑素沉积,同时伴基底层色素减退,细胞空泡化和变性,但有些病例基底层细胞内可有大量黑素。
诊断及鉴别
女婴有大疱和线状结节,或大疱和疣状损害合并出现,有特征性的色素沉着斑点出现容易诊断。应该与大疱表皮松解症及儿童期大疱性类天疱疮相鉴别。与Franceschetti-Jadasson综合征的区别是,后者的色素沉着呈网状分布而不是斑点状和涡轮状,并且无牙齿异常和眼部损害。与脱色性色素失禁症(无色素性色素失禁症或Ito黑素减少症)的区别在于该病是常染色体显性遗传病,无水疱期和疣状期,皮损为色素减退,并有中枢神经系统异常的高发生率。
预防及治疗
目前无特殊治疗。用红宝石激光治疗婴幼儿色素沉着无必要,并且可能加重病情。通常皮损在2岁以后开始逐渐消退,到成年期除有一些原有并发症外,几乎无任何不适。
