
拓扑异构酶(Topo)Ⅱ与MDR
近年来研究发现,一些对鬼臼毒素及其人工半合成衍生物Vp-16、VM-26、丝裂蒽酮、19羟基玫瑰树碱及安吖啶(AMSA)产生耐药性的细胞株,呈现一些非典型的多药耐药性现象,主要表现为:
- 对多种天然来源的抗肿瘤药物呈现耐药性,但对长春碱类生物碱则不产生交叉耐药性;
- 维拉帕米等膜活性物质并不能纠正其耐药性,因而维拉帕米等钙拮抗剂不能逆转这种类型的多药耐药性;
- 抗肿瘤药物在胞内的积聚及滞留没有变化,其胞内浓度未见下降;
- mdrl基因及Pgp的表达未见增加;
- 拓扑异构酶TopoⅡ酶的量及活性均有下降。因为与拓扑异构酶TopoⅡ有关的MDR同典型的由Pgp介导的MDR有诸多不同点,把这类MDR称为非典型性MDR(atypical MDR,at-MDR)。
拓扑异构酶是存在于真核细胞中的一种核酶,它可引起DNA的二维及三维结构改变,在DNA复制、转录、染色体分离及DNA重组中起重要作用。DNA拓扑异构酶有两种同工酶:TopoⅠ使DNA单链断裂并瞬间连接,无需ATP提供能量;TopoⅡ可使双链断裂并瞬间连接,需ATP提供能量。二者在维持核酸的正常结构及功能中起重要作用。许多抗肿瘤药物能与Topo结合后,形成永久性易裂性药物-酶-DNA复合物,从而阻止Topo的DNA断裂-再连接反应,导致DNA链断裂,进而影响蛋白质形成,促使肿瘤细胞死亡,或诱导基因的变化引起细胞凋亡,这亦是上述抗肿瘤药物的主要抗肿瘤作用机制。在目前常用的抗肿瘤药物中,喜树碱及其新的衍生物拓扑特肯和CPT-11是TopoⅠ抑制剂,鬼臼毒素类衍生物VP-16和VM-26是TopoⅡ抑制剂,胺苯吖啶和典型的蒽环类抗癌药(ADM和DNR)则为DNA嵌入剂,而长春碱类不作用于DNA。
目前有关拓扑异构酶与肿瘤MDR的研究主要集中在TopoⅡ,而其改变导致MDR的主要机制为:
- 细胞内TopoⅡ含量下降:人小细胞肺癌耐药细胞GLC4/ADM中未见Pgp过度表达,维拉帕米亦不能逆转其对阿霉素的耐药性,该细胞对VP-16、VM-26、mAMSA及米托蒽醌有交叉耐药,从该细胞系中提取TopoⅡ发现,DNA双链断裂活性较敏感细胞下降2~3倍,但TopoⅡ活性则未见明显改变;
- TopoⅡ基因点突变:某些TopoⅡ抑制剂如胺苯吖啶和VP-16本身为致突变剂,药物引起编码TopoⅡ的DNA发生点突变,导致TopoⅡ相应氨基酸序列改变,酶活性下降,从而改变了酶对抗癌药物的敏感性;
- TopoⅡ的磷酸化:研究发现TopoⅡ的磷酸化堆砌活性具有重要调节作用,可直接影响对抗癌药物的敏感性,Ritke等(1994)发现耐VP-16的K562耐药细胞TopoⅡ磷酸化水平比亲代敏感细胞低2. 6倍,VP-16诱导的DNA损伤减少,TopoⅡ共价结合复合物解离加速,提示该细胞耐药性与TopoⅡ磷酸化水平下降有关;
- Topo亚型的转换:最近研究发现TopoⅡ有两种亚型TopoⅡα和TopoⅡβ,二者具有不同的生物学特性、结构、功能和调节机制及对化疗药物的敏感性,如耐mAMSA的P388细胞耐药性与TopoⅡα减少有关,而耐米托蒽酮的HL60细胞则主要与TopoⅡβ表达下降有关。
关于TopoⅡ含量和活性的改变在临床耐药性方面的意义,目前尚不清楚,仅在一部分患者中观察到酶水平与药物敏感性有明确的关系。此外,近年来TopoⅠ与MDR的关系也越来越引起人们的重视。
细胞内解毒系统与MDR
细胞内解毒系统包括谷胱甘肽(glutathione,GSH)及相关酶,即谷胱甘肽-硫转移酶(glutathione-S-transferase,GST)和谷胱甘肽过氧化物酶(glutathione peroxidase,GSH-pX)及超氧化物歧化酶(superoxide dismutase,SOD)。
GSH是细胞内主要的非蛋白质巯基化合物,有还原型和氧化型两种,其主要功能是保护氧化剂对巯基的破坏,保护细胞膜中含巯基蛋白质和巯基酶不被氧化。抗肿瘤药物多属亲电子化合物,在GST等相关酶催化下与GSH结合形成GS-X复合物,然后被转运到胞外。GST属Ⅱ相药物代谢酶,是一种多功能酶,根据其等电位不同可分为GSTα、μ和π三种亚型,GST不仅可催化亲电子化合物与GSH结合,而且GST自身可与亲脂性细胞毒药物结合,增加其水溶性,促进其排泄,从而可保护细胞免受细胞毒药物的伤害,同时抗肿瘤作用亦因之下降。
人们在研究肿瘤MDR时发现细胞内GSH和(或)GST表达往往增加,提示GSH/GST与肿瘤MDR有一定关系,进一步研究发现:
- GST可代谢美法仑、卡莫司汀及丝裂蒽酮等抗肿瘤药物;对苯丁酸氮芥呈耐药性的CHO细胞,对卡莫司汀呈耐药性的大鼠胶质瘤细胞以及对顺铂呈耐药性的细胞株,均呈现GST高表达;
- 采用免疫组化研究显示,结肠癌和非小细胞肺癌组织中GSTπ表达较小细胞肺癌为高,提示GSTπ可能是耐药性标志之一;
- 用谷酰胺半胱氨酸合成酶抑制剂丁硫氨酸亚砜胺(BSO)使细胞内GSH水平下降,肿瘤细胞的敏感性也随之恢复;
- GST抑制剂如依他尼酸(ethacrynic acid)和PGI抑制剂,也可明显增加一部分MDR细胞对化疗药物敏感性;
- GST基因或GST cDNA转移到敏感细胞后,对特异性药物的耐药性可能提高。
虽然体外药物选择性耐药细胞系证实与GSH/ GST,特别是与GSTπ增加有关。但由于GSH/GST解毒系统广泛存在于人体各种器官中,故治疗前GSH/GST增加尚不能完全肯定为原发性耐药,但作为MDR家族的一员可能参与MDR的形成。有研究显示mdr1和GSTπ共表达与耐药之间存在明显相关性,提示mdr1为其主要耐药机制,GST解毒机制可能作为辅助因素参与MDR;而GSTπ与mdr1表达不一致者,可能与肿瘤类型不同有关,因此GST可能为某些肿瘤的标志物。最近研究表明MRP是一种GS-X泵,除能转运GSH耦联复合物,亦能调控某些天然来源的抗肿瘤药物,如长春碱类、鬼臼毒素类和蒽环类抗癌药物,这可能是GSH/GST导致MDR的另一耐药机制。目前有关GSH/GST临床表达与耐药、疗效及预后关系的研究尚不多见。
此外,MDR的产生可能与谷胱甘肽过氧化物酶活性升高也有关,GSH也可在GSH-PX的催化下,促使H2O2及氧自由基还原为H2O,清除自由基和超氧化物,而有些抗肿瘤药物能提供氧自由基、H2O2及OH•活性氧直接损伤DNA而产生细胞毒作用。
蛋白激酶C与MDR
蛋白激酶C(protein kinase C,PKC)是一种磷脂依赖性丝氨酸/苏氨酸蛋白激酶家族,明确已发现至少有12个同工酶亚型,广泛分布于不同组织及其不同的细胞区域。其结构主要含有两个功能单位,即与磷脂、二酯酰甘油(DAG)及佛波酯(TPA)结合的疏水性调节单位,和与ATP及底物结合的催化单位。
PKC活化在机体对外界刺激产生的信号传导通路中起着十分重要的作用,它把许多胞外信号,如生长因子、激素、细胞脂多糖及神经递质等,在细胞膜上经受体介导的第二信使传递入核,从而使细胞对外界产生一系列反应,调节着细胞的基因表达、代谢、增殖、分化和凋亡,是细胞信号通路的中心分子。
近年来的研究表明肿瘤MDR的形成也与PKC活化有关,其证据主要有:
- 在耐药细胞中的PKC活性远远高于敏感细胞;
- 肿瘤细胞暴露于PKC激动剂TPA,可激活Pgp的磷酸化,导致细胞对药物的耐受;
- 许多MDR逆转剂如吩噻嗪、三氟拉嗪和他莫昔芬等,能够降低PKC活性,恢复对化疗药物的敏感性。
PKC有多种亚型,哪一个PKC亚型与MDR有关?有人比较了敏感细胞系和MDR细胞系中PKC的亚型分布,发现PKC-γ仅存在于HL60/ADM耐药细胞系中,与敏感细胞相比,MDR细胞中PKC-β含量略低,PKC-α含量则无某些差别,提示PKC-γ可能与MDR有关。但在P388/ADM耐药细胞系中,PKC-α和PKC-β活性均较相应的敏感细胞为高。将编码PKC-α及PKC-β亚型的cDNA转染敏感细胞后,亦可获得MDR细胞,亦直接证明了PKC-α和PKC-β在MDR产生中的启动作用,可见PKC各亚型在产生MDR中的作用尚存在着异议,有待进一步研究。
关于PKC在MDR发生、发展中的作用机制目前尚未完全阐明,但普遍认为PKC是通过诱导mdr1基因过度表达和加速Pgp的磷酸化而导致MDR的发生与发展的。极有兴趣的是,PKC活性增高并不能增加非MDR药物Ara-C的细胞毒作用,相反用PKC激动剂使PKC活性提高4~5倍后,耐药细胞对顺铂的敏感性提高9倍,进一步说明非MDR药物的耐药性与mdr1基因及Pgp无关。
可溶性耐药相关钙结合蛋白Sorcin 与MDR
可溶性耐药相关钙结合蛋白Sorcin是20世纪80年代从中国仓鼠耐药细胞发现的胞质蛋白。人与仓鼠sorcin基因的同源物与编码P-糖蛋白基因(MDR1,MDR3)相似,位于7号染色体。在一株人卵巢癌细胞株中发现class 4、5和6的基因与MDR1 和MDR3一起扩增,表明人的MDR辖区(domain)的全部结构与仓鼠和小鼠由扩增而引起MDR辖区的结构是相似的。它不仅包括编码P-糖蛋白的基因,而且编码至少5个其他不相关的基因,其中之一,由class 4基因编码的钙结合蛋白sorcin是一种小的胞质蛋白,它在许多MDR细胞中过度表达。它有与钙蛋白酶调节性轻链相似的序列,包括在钙调蛋白中钙结合类型的环。由于用sorcin基因转染细胞没有获得耐药性,加之sorcin在耐药细胞株的表达会随选择时间延长而消失,故又称“过客基因”(passenger gene)。所以sorcin与耐药关系就没有得到进一步的研究。
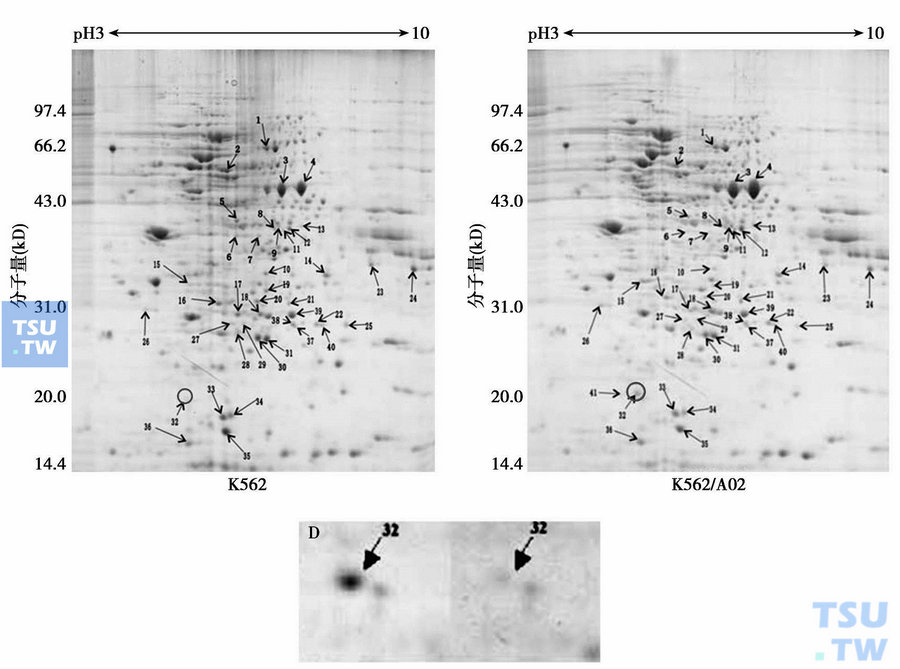
双向凝胶电泳法分析多药耐药细胞株K562/A02和其亲代细胞株K562中基因表达差异,32号为Sorcin基因表达蛋白
我们用c- DNA微阵列芯片检测1176个人类基因,比较敏感K562细胞和它的耐药细胞K562/A02的基因表达发现,在K562/A02细胞过度表达的基因中,最强的就是sorcin,并且它的蛋白组学研究中得以进一步证实(上图)。这表明它与mdr1在K562/ A02细胞共同表达,表达强而稳定,并非“过客基因”。这就促使我们进一步研究它与mdr1在白血病MDR中的意义。我们在临床用RT- PCR方法检测95例急性白血病患者和27例非白血病患者和健康人的sorcin基因表达水平。研究结果发现,白血病患者sorcin基因表达高于正常对照组,差异有高度统计学意义;急性髓性白血病(AML)复发难治组高于初诊组和完全缓解组;临床耐药组sorcin基因阳性表达显著高于非临床耐药组,差异有显著统计学意义;sorcin 与mdr1表达高度相关,它们在耐药患者的共表达达92%。因此,sorcin既可作为检测AML临床耐药和判断预后的独立指标又可作为与mdr1共表达指标。这是我们首次发现sorcin的临床意义,进一步的研究正在进行。
白血病sorcin基因表达与临床耐药的关系
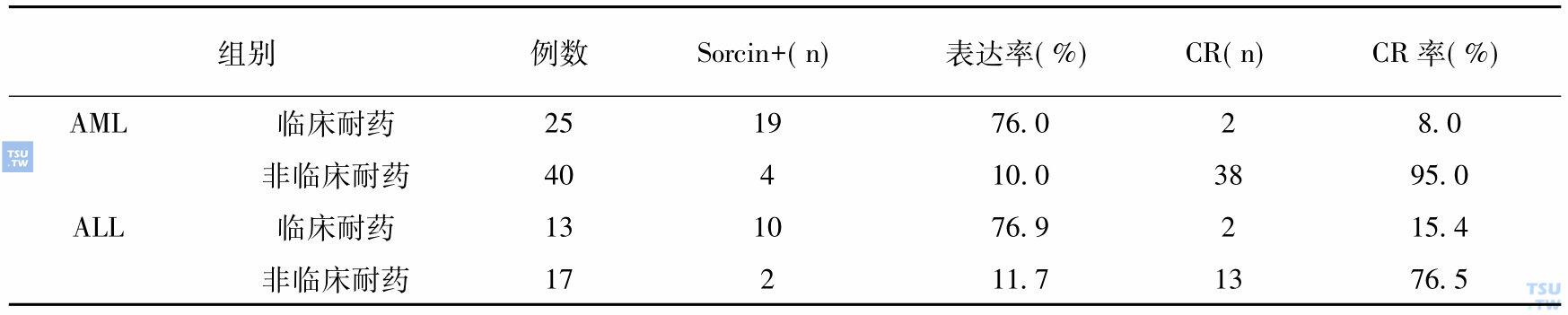
注释:CR表示完全缓解
sorcin基因与mdr1/Pgp基因共表达的临床意义
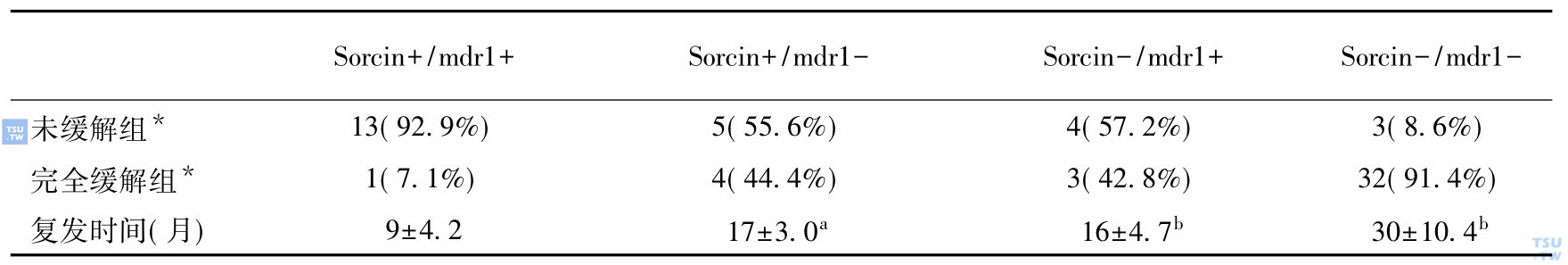
注释:a:Sorcin+/mdr1-组与Sorcin+/mdr1+比较,P<0. 005
b:标注组与Sorcin+/mdr1+比较,P<0. 001,*单位为例数
c:()中数字为NR%
