
目前认为,2型糖尿病是一种遗传和环境因素共同作用而形成的多基因遗传性复杂疾病,其特征为胰岛素抵抗、胰岛素分泌不足和肝糖输出增多。调节代谢和胰岛素抵抗的新途径有FGF21、脂联素和PPARr系统。FGF19、FGF21 和FGF23是体内矿物质和其他物质代谢调节的关键因子。α-klotho-1(α-K1)、FGF23、1,25-(OH)2D和PTH形成矿物质调节网络,而FGF19和胆酸调节体内酸碱和胆固醇代谢。在脂肪组织中,FGF21具有klotho依赖和非klotho依赖的两条途径,调节能量代谢。
大多数2型糖尿病为多个基因和多种环境因素共同参与并相互作用的多基因多环境因素复杂病(complex disease),一般有以下特点:
- 参与发病的基因多,但各参与基因的作用程度不同;起主要作用者为主效基因(major gene or master gene),作用较小者为次要基因(minor gene),即各个基因对糖代谢的影响程度与效果不同,各基因间可呈正性或负性交互作用;
- 不同患者致病易感基因的种类不同,非糖尿病者也可有致病易感基因,但负荷量较少;
- 各易感基因分别作用于糖代谢的不同环节。这些特点赋予2型糖尿病的异质性,给遗传学病因研究带来极大障碍。
2型糖尿病具有多基因遗传背景
胰岛素抵抗和胰岛β细胞功能缺陷(胰岛素分泌不足)是2型糖尿病的基本特征,研究导致两方面缺陷的候选基因功能和致病原理,是探讨2型糖尿病发病机制的重要途径。2007年以来,糖尿病的全基因组关联分析研究结果不仅肯定了PPARγ、KCNJ11 和TCF7L2基因与2型糖尿病的相关性,还发现了多个新的与2型糖尿病相关的基因。到目前为止,随着多个GWAS研究结果的陆续发表和对多个GWAS研究数据的综合分析,人们已经发现了近40个新的2型糖尿病基因和数个和2型糖尿病相关性状如体重、血糖及HbA1c相关的基因,并发现TCF7L2基因的致病作用最大,但迄今尚未发现主效基因。2型糖尿病有明显的遗传易患性,并受到多种环境因素的影响,其发生的核心问题是胰岛素,胰岛素的主要功能是促进脂肪分解、抑制肝糖输出以及增加肌肉组织对葡萄糖的摄取。当患者出现糖尿病的时候,一方面有β细胞功能紊乱,另一方面患者还可能存在不同程度的胰岛素抵抗,两者不同程度地影响胰岛素的功能(下图)。两方面的缺陷在不同的个体表现轻重不一。因而,2型糖尿病个体之间存在明显的异质性。
2型糖尿病的病因与发病机制;注:GLUT:葡萄糖转运蛋白;IRS:胰岛素受体底物。
遗传因素在2型糖尿病的病因中较l型糖尿病明显。同卵双胎患2型糖尿病一致率为90%,双亲中1人患2型糖尿病,其子女患病的风险率为5%~10%;父母皆患病的子女中,5%有糖尿病,12%有IGT。表现在:①家系调查发现2型糖尿病 38%的兄妹和1/3的后代有糖尿病或糖耐量异常。据报道,我国 25岁以上糖尿病患者群中糖尿病家族史阳性率为14%,正常人群是7.4%;糖尿病患者群中父亲和母亲糖尿病家族史阳性率无差异;有糖尿病家族史的糖尿病者发病年龄早,2/3均在54岁以前发病。起病早的2型糖尿病患者家族史较多见,40岁前起病的2型糖尿病患者的双亲及同胞的患病率明显高于40岁或以后起病者。张素华等对2型糖尿病和家系胰岛素分泌功能的研究发现2型糖尿病家系中,各成员均存在高胰岛素血症,一级亲属胰岛β细胞初期分泌功能代偿性增强,以维持正常的糖耐量;②孪生子患病一致率研究发现,2型糖尿病双胞胎中58%有糖尿病,追踪10年其余大部分人也发生糖尿病。同卵双生的双胞胎中,2型糖尿病的发病率可达70%~80%;③糖尿病患病率有明显的种族和地域差异,从患病率几近0的巴布亚新几内亚到患病率最高的美国亚利桑那州的Pima印第安人及西南太平洋密克罗尼西亚群岛的Nauru人。35岁以上的Pima印第安人中50%以上患2型糖尿病。生活方式现代化使这两种人2型糖尿病的患病率急剧增加。在年龄大于60岁的Caucasians白人人群中,2型糖尿病的患病率大约为10%。在年龄大于60岁的纯种Nauru人中,2型糖尿病的患病率大约为83%,在混血儿中则大约为17%。
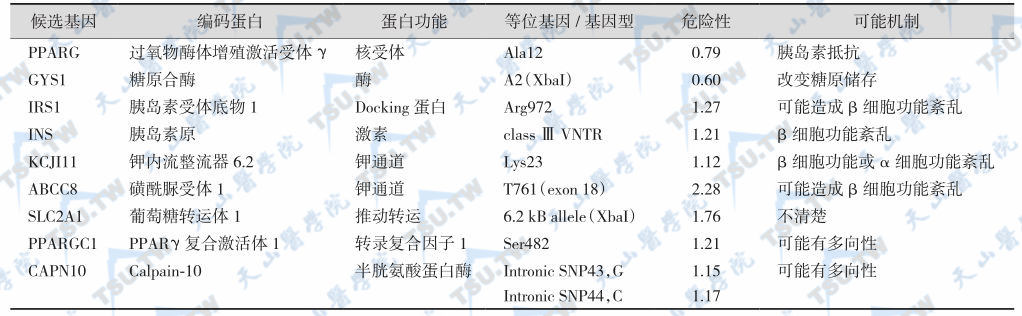
参与发病的遗传因素不止1个,可能多达数十个,已经发现许多与2型糖尿病相关的候选基因(下表)。每个基因参与发病的作用大小不一,大多数基因的作用很小,甚至是微效的,称之为次效基因(minor gene),但有1个或几个基因的作用呈明显的主效效应,为主效基因(major gene)。每个基因只赋予个体对2型糖尿病某种程度的易患性。
与2型糖尿病相关的候选基因和多肽
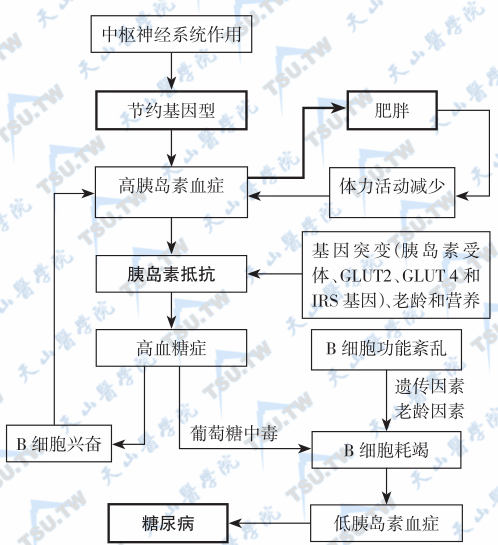
遗传因素参与2型糖尿病发病的机制:①“节俭基因型”假说提出,人类进化过程中所选择的“节俭基因型”,有利于食物充足时促进脂肪堆积和能量储存,以供经常发生的天灾饥荒时食物短缺时耗用。人类中具有在进食后能较多地将食物能量以脂肪形式储存起来的个体,就较易耐受长期饥饿而生存下来。通过自然选择,这种有“节俭基因型”的个体在人类进化中,有利于在逆境中生存而被保留下来。但是到了食品供应充足的现代社会,有“节俭基因型”个体就易出现肥胖、胰岛素抵抗和糖尿病,也就是说在体力活动减少和热量供应充足的情况下,节俭基因成了肥胖和2型糖尿病的易感基因。②“共同土壤”假设认为这些疾病有各自不同的遗传和环境因素参与发病,但还可能有共同的遗传及环境因素基础。③糖尿病并发症,尤其是糖尿病肾病和糖尿病视网膜病的发生也存在有别于糖尿病的遗传因素的参与。糖尿病肾病和视网膜病变代表糖尿病微血管病变,存在明显的家族聚集倾向,家族内孪生子、同胞及亲属患者之间上述并发症发生的一致率高。
诱发2型糖尿病的因素
流行病学研究表明,肥胖、高热卡饮食、体力活动不足和增龄是2型糖尿病的主要环境因素,有高血压、血脂谱紊乱、IGT 或IFG者的2型糖尿病患病风险增加。在这些环境因素中,肥胖居于中心地位。
肥胖
在2型糖尿病中,肥胖被认为是重要的环境因素。具有2型糖尿病遗传易患性的个体中,肥胖有使2型糖尿病呈现的作用。而且,肥胖的2型糖尿病体重减轻后,糖尿病的临床症状可减轻甚至糖耐量也可恢复正常,这是不争的事实。流行病学研究显示,肥胖和体力活动不足是2型糖尿病的重要危险因素;肥胖和超重是发展中国家糖尿病患病率急剧攀升的主要原因;肥胖患者存在高胰岛素血症和胰岛素抵抗,胰岛素调节外周组织对葡萄糖的利用率明显降低,周围组织对葡萄糖的氧化和利用障碍,胰岛素对肝糖生成的抑制作用降低,非酯化脂肪酸(FFA)升高;高水平的FFA可刺激β细胞分泌胰岛素增多而产生高胰岛素血症,并损害胰岛β细胞功能;FFA可明显抑制β细胞对葡萄糖刺激的胰岛素分泌;FFA升高可能使胰岛β细胞中脂酰辅酶A升高,后者为三酰甘油(TG)合成的原料,胰岛β细胞中脂质的增加可能影响其分泌胰岛素的功能。肥胖患者存在明显的高胰岛素血症,高胰岛素血症降低胰岛素与受体的亲和力。亲和力降低,胰岛素的作用受阻,引发胰岛素抵抗,需要β细胞分泌和释放更多的胰岛素,又引发高胰岛素血症,如此呈糖代谢紊乱与β细胞功能不足的恶性循环,最终导致β细胞功能严重缺陷,引发2型糖尿病。
一:中心型肥胖
在肥胖中,中心型肥胖是促发2型糖尿病的一个重要因素。中心型肥胖即腹型肥胖,腹内脂肪与全身脂肪的比值升高,临床用腰、髋比值(WHR)估计(见第5篇第31章第248节)。内脏脂肪蓄积引发胰岛素介导的葡萄糖清除率明显降低,促进胰岛素抵抗,导致脂代谢紊乱和高血压。体重除受遗传因素(如ob基因和PPARγ基因等)的控制外,还受环境因素的影响。Hales等用节约基因型(thrifty genotype)假说来解释这种现象,该假说认为,长期生活在食物匮乏条件下的人群高度表达有利于生存的节约基因,将体内的剩余营养物质以脂肪形式贮存下来,供饥荒时使用;当这些人群进入体力活动少和热卡供给充足过剩的现代社会后,节约基因不能及时适应生活方式的快速改变,转变成肥胖和2型糖尿病的易感基因。当摄入高热量、饮食结构不合理(高脂肪、高蛋白和低碳水化合物)和体力活动不足时,易导致肥胖,肥胖再降低胰岛素敏感性,促进糖尿病的发生。食物摄入过量和缺少运动是导致肥胖的主要环境因素,特别是在有“节俭基因型”的个体。幼年时期生活在贫困地区的人们,在较富裕的生活环境中特别易发生肥胖和IGT。2010年ADA会议上我国研究者报道,中国在20世纪50年代晚期至20世纪60年代早期经历了分布广且严重的饥荒,造成数百万人死亡。1959~1961年是饥荒最严重,死亡率最高的时期。调查出生前和儿童时期经历的饥荒与成人后高血糖和2型糖尿病风险之间的关联。结果发现:胎儿时期经历严重饥荒增加成人后的高血糖风险,后期营养过盛的环境令这一关联恶化。
二:棕色组织
患2型糖尿病的日本人和中国人30%有肥胖,北美人60%~70%存在肥胖,Pima印第安人和南太平洋的Nauru和Samoa人几乎全部伴有肥胖。流行病学调查显示,肥胖者的外周组织胰岛素受体数目减少、葡萄糖氧化利用或非氧化利用障碍、胰岛素对肝糖输出的抑制作用降低和游离脂肪酸代谢增高均可影响葡萄糖的利用,需分泌更多的胰岛素代偿缺陷。虽然肥胖者均存在胰岛素抵抗,但内脏型肥胖较外周肥胖、脂肪细胞体积增大较数目增多更易发生胰岛素抵抗。在遗传背景的影响下,长期而严重的胰岛素抵抗最终导致β细胞功能衰竭。
肥胖具有强烈的遗传背景,食欲、食量和摄食选择均受遗传因素的影响。当机体摄食或受寒冷刺激时,棕色脂肪分解产热,向体外散发热量。肥胖者的棕色脂肪细胞功能低下,进餐后的摄食诱导产热占总能量消耗的9%,而体瘦者占15%。体脂含量、体脂分布和脂肪细胞功能也主要由遗传因素决定,现已确定了数种肥胖相关基因及其相关蛋白。β3肾上腺素能受体(β3AR)活性下降对内脏型肥胖的形成有重要作用,内脏脂肪中β3AR的活性较皮下脂肪高,儿茶酚胺与β3AR结合后启动蛋白激酶磷酸化,促进脂肪分解并发挥产热作用。β3AR活性降低时,通过减少棕色脂肪的产热作用而使白色脂肪分解减慢,造成脂肪蓄积与肥胖。
目前已经鉴定了数十种脂肪细胞因子,至少其中的部分因子与肥胖和2型糖尿病相关:①脂肪细胞分化和增殖至少受转录因子CAAT/增强子结合蛋白(CAAT/enhancer binding protein,C/EBP)和过氧化物酶增殖体活化受体-γ(peroxisome proliferator-activated Receptor-γ,PPAR-γ)的调节,PPAR-γ基因突变可导致严重肥胖;②脂肪细胞合成和分泌瘦素(leptin),其与下丘脑受体结合后抑制神经肽Y(neuropeptide Y,NPY)基因转录,使下丘脑弓状核神经元合成的NPY减少,抑制食欲,减少热量摄入,提高机体代谢率,减少脂肪堆积,故瘦素缺乏或抵抗是肥胖的另一个原因;③食欲素orexin有食欲调节作用,而orexin A是拮抗瘦素的主要因子;④内脏脂肪素(visfatin)可结合并激活胰岛素受体,模拟胰岛素作用,降低血糖,并促进脂肪细胞分化、合成及积聚;⑤visfatin、抵抗素(resistin)与肥胖及胰岛素抵抗的关系有待进一步研究。
三:脂毒性
脂毒性(lipotoxicity)在2型糖尿病及其并发症的发病中有重要作用。血脂紊乱时,血浆游离脂肪酸(free fatty acid,FFA)长期升高导致脂肪酸和甘油三酯在非脂肪组织(胰岛β细胞、骨骼肌、心脏和肝脏等)沉积。脂肪酸特别容易发生氧化损伤,形成高反应性的脂质过氧化物(活性氧,reactive oxygen species,ROS),导致胰岛素抵抗、2型糖尿病及其慢性并发症。
ROS具细胞毒性,可导致蛋白质和DNA的自由基损伤,其后果为:①促进胰岛β细胞凋亡;②抑制骨骼肌胰岛素信号传导和GLUT4的生成与转位;③激活丝氨酸激酶抑制蛋白激酶β(IKK-β)/NF-κB旁路,介导胰岛素抵抗;④引起心脏功能障碍和脂肪肝。
过多脂肪异位储积(ectopic fat storage)于肝脏、肌肉、脾脏、胰腺和其他内脏器官。在脂肪细胞因子和内分泌激素的作用下,脂解增加,血甘油三酯升高,肝游离脂肪酸释放增多,最终引起胰岛素抵抗和2型糖尿病。内脏脂肪蓄积引发胰岛素介导的葡萄糖清除率降低,促进胰岛素抵抗,导致脂代谢紊乱、高血压、糖耐量低减或糖尿病。
不合理饮食
高脂肪膳食与肥胖、血糖水平和糖尿病的患病率密切相关,富含纤维和植物蛋白的膳食有预防糖尿病的作用,食糖并不增加糖尿病的患病率。脂肪摄入过多是2型糖尿病的重要环境因素之一。食物中不同类型的脂肪酸对胰岛素抵抗产生不同的影响。脂肪酸是构成人体脂肪和类脂(磷脂、糖脂和类固醇等)的基本物质,根据碳氢链中双键的有无,将脂肪酸分为不含键的饱和脂肪酸(SFA)和含有双键的不饱和脂肪酸;不饱和脂肪酸又可根据其所含双键的多少分为仅含1个双键的单不饱和脂肪酸(MuFA)和含1个以上双键的多不饱和脂肪酸(PuFA);PuFA又可根据最靠近碳原子双键的位置进一步分为ω-3(omega-3)和ω-6(omega-6)等系列脂肪酸。所谓ω-3系列PuFA就是指从脂肪酸碳链甲基端算起,第1个双键出现在第3位碳原子上的PuFA。食物中脂肪主要指各种植物油和动物脂肪。食物中SFA主要存在于动物脂肪、肉及乳脂中,植物油中含量极少。MuFA主要为油酸(18碳1烯酸),在橄榄油中含量最多(84%)。
ω-6系列PuFA(简称ω-6脂肪酸)富含于植物油中。主要成分为亚油酸(18碳乙烯酸)和由此转化而来的花生四烯酸(AA,20碳4烯酸)。ω-3系列PuFA(简称ω-3脂肪酸)主要成分为亚麻酸(18碳3烯酸)、EPA(20碳5烯酸)和DHA(20碳6烯酸)。亚麻酸主要存在于亚麻油中(高达50%),因其独特的气味难为食用者接受,因此,它不是人类亚麻酸摄入的主要来源,其他植物油如豆油和玉米油等含程度不同的亚麻酸。除亚麻酸在体内能转化少量EPA和DHH外,EPA和DHH主要来源于深海鱼类(鱼油和鱼内脏中)。多因素分析发现空腹胰岛素水平与脂肪和SFA摄入量呈正相关,与MuFA和PuFA摄入无相关。提示饮食中合理减少脂肪和SFA摄入将有助于预防糖尿病。美国ADA推荐:饮食中脂肪酸摄入标准是脂肪供能在总热能中应低于30%,其中SFA<10%,PuFA<10%,MuFA<10%~15%。
食用水溶性纤维可在小肠表面形成一种高黏性液体,包被糖类,从而对肠道的消化酶形成屏障,延缓胃排空,从而延缓糖的吸收。食用水溶性纤维可被肠道菌群水解,在肠道中形成乙酸盐和丙酸盐,这些短链脂肪酸可吸收入门静脉,并在肝脏刺激糖酵解,抑制糖异生,促进骨骼肌葡萄糖转运蛋白4(GLUT4)表达。此外,水溶性纤维尚可减少胃肠激素的分泌,而胃肠激素刺激胰岛分泌胰岛素,因此,高纤维饮食可改善胰岛素抵抗和降低血糖。高果糖摄取可以增加血浆C肽浓度,每日用66%的果糖喂养大鼠2周,其骨骼肌和肝脏中的胰岛素受体数和胰岛素受体mRNA比标准食物喂养大鼠明显降低,而血压和血浆TG明显增加。食物中锌和铬的缺乏,可使糖耐量减低,2型糖尿病的发病率增加。酗酒也可引发糖尿病。
体力活动不足
流行病学调查发现,强体力劳动者发生2型糖尿病者远低于轻体力劳动或脑力劳动者。运动可改善胰岛素敏感性。用葡萄糖钳夹技术研究表明,即使运动不伴体重下降,血浆胰岛素水平和胰岛素释放面积也降低,葡萄糖清除率增加。运动可使胰岛素与其受体的结合增加,从而改善胰岛素抵抗和胰岛素作用的敏感性,而且适当的运动还有利于减轻体重,改善脂质代谢。
低体重儿
“成年疾病的胎儿(早期)来源假说”(fetal or early origins of adult disease hypothesis)认为,环境因素或营养因素作用于生命体早期,编制出疾病状况(如高血压、胰岛素抵抗、肥胖和代谢综合征等)。流行病学和实验动物证实,宫内生长迟缓(intrauterine growth retardation,IUGR)的低体重儿与成年2型糖尿病胰岛β细胞功能受损和胰岛素抵抗相关。
肠促胰素分泌缺陷
肠促胰素(incretin hormones)是一类肠源性激素,包括胰高血糖素样肽1(GLP-1)和葡萄糖依赖性促胰岛素多肽(GIP)等。由胃肠道L细胞生成的GLP-1和由K细胞生成的GIP都具有葡萄糖浓度依赖性胰岛素分泌的刺激作用(肠促胰素效应),其作用途径是1型味觉受体(taste type 1 receptor,T1R),其配体是甜蛋白(sweet protein)brazzein。GLP-1的降糖效应至少来自以下4个方面:①促进胰岛素分泌,增加胰岛素合成,减少β细胞凋亡并促进其增殖,增加β细胞数量;②减少α细胞的胰高血糖素分泌;③作用于脂肪、肌肉和肝脏,增加葡萄糖摄取,减少肝糖输出,协同胰岛素降低血糖;④作用于中枢的食欲控制系统,增加饱感,延缓胃排空,减少摄食,间接降低血糖。GLP-1作用于血糖去路和来源多个靶点的降血糖效应是独特的。但是,2型糖尿病患者口服与静脉葡萄糖刺激下的胰岛素分泌差值显著降低,即肠促胰素效应明显减弱,其主要原因是肠促胰素分泌减少和作用缺陷。
胰岛素抵抗存在于多个环节
胰岛素抵抗(insulin resistance,IR)在2型糖尿病发生中处于核心地位(下图)。IR和β细胞分泌缺陷是2型糖尿病发病机制的两个主要环节。IR是2型糖尿病的特征之一,在出现临床高血糖前就已经存在。IR的概念是机体对一定量(一定浓度)胰岛素的生物效应减低,主要指机体胰岛素介导的葡萄糖摄取和代谢能力减低,包括胰岛素的敏感性下降和反应性下降。胰岛素在调节机体葡萄糖稳态中起关键作用。其主要的效应器官是肝脏、骨骼肌及脂肪组织。胰岛素主要的生理效应包括其介导葡萄糖的摄取及处置(糖的氧化及贮存)、促进蛋白质合成、促进脂肪合成、抑制糖异生、抑制脂肪分解及酮体生成等。IR可发生于组织器官水平(骨骼肌、脂肪、肝脏和血管内皮),也发生于亚细胞及分子水平(胰岛素受体前、受体和受体后)。
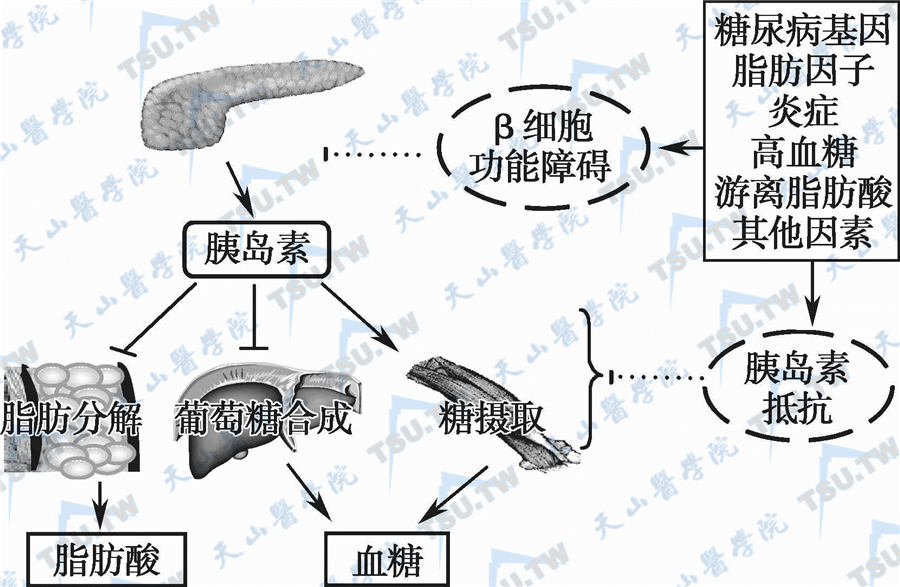
胰岛素抵抗在2型糖尿病发生中的地位
胰岛素受体前抵抗
引起受体前胰岛素抵抗的原因有胰岛素分子结构异常、胰岛素抗体、胰岛素降解加速和拮抗激素增多等。胰岛素基因突变可产生结构异常的胰岛素,使胰岛素的生物活性下降或丧失,如Chicago胰岛素(PheB25Leu)、Los Angeles胰岛素(PheB24Ser)、Wakayma胰岛素(ValA3Leu)、Providence胰岛素(HisB10Asp)以及Tokyo胰岛素原(Arg65His)。内源性或外源性胰岛素抗体形成,可干扰胰岛素与受体的正常结合。后者常见于注射纯度低的动物胰岛素时,抗体形成的高峰时期是注射胰岛素后3~4个月。胰岛素抗体是否影响胰岛素发挥其正常功能与抗体的胰岛素识别位点密切相关。在胰岛素抗体中,只有当抗体的胰岛素识别位点与胰岛素的受体结合区域相重叠时,才会有阻断胰岛素的作用;在携带胰岛素抗体的糖尿病患者中,胰岛素抗体的胰岛素识别位点对最终是否发生胰岛素抵抗起重要作用。胰岛素受体前抵抗还可由于胰岛素降解加速引起。一些药物如糖皮质激素、生长激素(GH)、苯妥英钠、INF-γ、INF-α等及其他应激激素分泌过多(如感染、创伤、手术、酮症酸中毒、Cushing综合征和肢端肥大症等)均可导致受体前抵抗。
胰岛素受体缺陷
胰岛素受体缺陷包括胰岛素受体功能与结构的异常。其功能异常包括胰岛素受体数目减少以及亲和力下降导致与胰岛素结合减少;其结构异常多为胰岛素受体基因(IRG)突变,致使受体功能完全丧失或部分丧失。1988年以来,已发现50余个突变位点,按其对受体功能影响的不同可分为5类:
- Ⅰ类抵抗:IRG的外显子2、内含子4和外显子5拼接点的无义突变所导致的胰岛素受体合成障碍。临床上见于婴儿妖精症,为严重的IR,婴儿罕见存活至1岁以上。
- Ⅱ类抵抗:受体蛋白翻译后加工和分子折叠障碍,其结果使受体不能从细胞的粗面内质网及高尔基体转位至细胞膜,故而膜受体数目减少,其突变点主要在α亚基N端以Gly为中心的重复序列处。
- Ⅲ类抵抗:为受体亲和力下降,胰岛素与其受体的结合降低。突变点有3处,均在膜外区域(Asn15Lys、Arg735Ser及Ser323Leu)。
- Ⅳ类抵抗:受体β亚基酪氨酸激酶活性降低,导致β亚基自身磷酸化作用障碍,因而穿膜信号传导障碍,已发现突变基因位点10余个。
- Ⅴ类抵抗:基因突变导致受体降解加速。突变位点在α亚基Lys460Gln及Asn462Ser处。但是,以上所述的胰岛素受体缺陷所致的糖尿病均属于特殊糖尿病类型,通常的2型糖尿病与胰岛素受体缺陷无明显关系。
将小鼠不同组织的胰岛素受体敲除发现,敲除肝胰岛素受体小鼠表现出严重的胰岛素抵抗、肝功能受损和糖耐受异常;在肌肉组织敲除胰岛素受体,小鼠表现为中等度的肥胖,没有胰岛素抵抗和糖耐量受损;在脂肪组织敲除胰岛素受体,则表现为消瘦和寿命延长,没有糖耐量受损;在神经细胞敲除胰岛素受体,小鼠表现为多食、不育和肥胖,没有糖耐量受损;在胰岛β细胞敲除胰岛素受体,表现为胰岛素分泌缺陷,有糖耐量受损。这主要与胰岛素在不同组织器官的作用存在差别有关。
胰岛素受体后缺陷
系指胰岛素与受体结合后信号向细胞内传递所引起的一系列代谢过程,即所谓胰岛素受体的“下游事件”,包括信号传递和放大,蛋白质-蛋白质交联反应,磷酸化与脱磷酸化以及酶促级联反应等多种效应的异常。
一:葡萄糖转运蛋白异常
肌肉和脂肪细胞对胰岛素刺激的葡萄糖摄取主要通过对胰岛素敏感的GLUT4来进行。在基础状态下,细胞表面GLUT4很少,在胰岛素刺激下,胰岛素受体酪氨酸磷酸化信号的内传使胰岛素受体底物-1 (IRS-1)磷酸化,从而活化磷脂酰肌醇-3-激酶(PI3-K)激酶,触发富含GLUT4的小泡以胞吐形式由内核体(endosome)经由高尔基复合体向细胞表面转位,因而细胞表面GLUT4增多,组织对葡萄糖摄取增加。当GLUT4基因突变时,GLUT4合成及转位均受阻。在2型糖尿病、肥胖症或高血压中,均发现有GLUT4募集及转位障碍,从而使肌细胞的葡萄糖摄取明显减少。GLUT2合成异常可造成肝摄取葡萄糖减少,肝胰岛素抵抗和β细胞对葡萄糖感受性降低,胰岛素分泌减少。
二:细胞内葡萄糖磷酸化障碍
研究证明,非肥胖2型糖尿病患者肌细胞内的葡萄糖6-磷酸(G-6-P)浓度明显降低,葡萄糖磷酸化的速率降低约85%,同时伴GLUT4转位的缺陷,即使GLUT4正常后,糖磷酸化异常仍未能恢复。导致葡萄糖磷酸化障碍的原因是己糖激酶Ⅱ (HKⅡ)活性降低。而此酶活性降低又受糖原合成酶及丙酮酸脱氢酶活性降低的影响。
三:线粒体氧化磷酸化(OXPHOS)障碍
OXPHOS障碍可致能量产生障碍和胰岛素刺激的糖原合成减少。
四:IRS-基因变异
正常情况下,胰岛素与受体结合后信号向细胞内传导,首先由IRS-1介导,IRS-1起着承前启后的作用。细胞内许多含SH2的蛋白质与IRS-1分子上磷酸化的酪氨酸残基结合,如PI3-K的85kD亚基与其结合后,可激活此酶的催化亚基(110kD)。这样经过许多酶促反应而使蛋白磷酸酶-1磷酸化(活化),其结果是与糖原代谢相关的两个关键酶(糖原合酶与磷酸化酶激酶)脱磷酸化。前者脱磷酸化使酶活化而刺激糖原合成;后者脱磷酸化则使其失活,从而抑制糖原分解,其净效应为糖原合成增多,血糖维持正常。若IRS-I基因(定位于2q36-37)突变,可使IRS-1酪氨酸磷酸化减弱,而丝氨酸磷酸化增强,则可产生IR。业已发现IRS-1基因有4种突变与2型糖尿病关联,它们分别是Ala513Pro、Gly819Arg、Gly972Arg及Arg1221Gys。目前已了解几种IRS丝氨酸激酶与胰岛素受体后信号传递有关,如有丝分裂原蛋白激酶(MAPK)、c-Jun-NH2末端激酶(JNK)、非经典蛋白激酶C(PKC)和PI3-K等。细胞因子信号抑制物(suppressor of cytokine signalling,SOCS)竞争性抑制IRS-1 酪氨酸磷酸化和减少IRS 与调节亚单位p85的结合导致胰岛素抵抗。新近的研究发现SOCS3也通过泛素(ubiquitin)介导的降解途径,加速IRS-1/2的降解。另外,在2型糖尿病患者还发现了几种IRS-1基因多态性较一般人群常见。研究得较多的是甘氨酸972精氨酸多态性,1项丹麦的研究观察到这种多态性频率在正常人为5.8%,而在2型糖尿病患者为10.7%。
内质网应激
内质网应激(endoplasmic reticulum stress)在糖尿病的病因中,内质网应激起了重要作用,尤其在β细胞凋亡和胰岛素抵抗中,内质网应激可能是最关键的环节。在β细胞中,蛋白的非折叠反应成分(components of the unfolded protein response,UPR)在生理条件下起着有利的调节作用,而在慢性应激时起着β细胞功能紊乱和凋亡的激发作用。β细胞的生理功能是在高血糖时,能敏感地分泌胰岛素;但在慢性高血糖和高脂肪酸的长期刺激下,β细胞变得十分脆弱,特别容易受损,使其成为细胞衰竭的重要因素。因此,在病理情况下,UPR转变成激发β细胞功能紊乱和凋亡前期的内质网应激反应(proapoptotic ER stress response)物。内质网应激还是联系肥胖和胰岛素抵抗的病理因子。实验发现,摄入高脂饮食的肥胖动物在肝脏出现内质网应激,并通过JNK 途径抑制胰岛素的信号传递。此外,内质网应激可引起以细胞因子(IL-1β和IFN-γ等)为介导的β细胞凋亡;而NO耗竭内质网中的储备钙,抑制内质网的钙摄取等又进一步加重内质网应激反应。
脂肪因子
目前研究发现,与IR有关的细胞因子有:FFA、肿瘤坏死因子-α(TNF-α)、IL-6、瘦素、脂联素、抵抗素、visfatin、IL-1、IL-1Rα、IL-8、IL-10、IL-18、单核细胞趋化因子(MCP-1)、单核细胞迁移抑制因子(MIF)、TGF-β、C反应蛋白(CRP)和肿瘤坏死因子受体(TNFR)等。其中备受关注的是TNF-α、瘦素、脂联素、抵抗素以及新近发现的内脏脂肪素(visfatin)。
一:FFA
2型糖尿病常存在脂代谢紊乱,FFA增多。FFA增多可引起IR,其机制可能与FFA抑制外周葡萄糖的利用和促进糖异生有关。FFA除对葡萄糖氧化途径有抑制作用外,对葡萄糖的非氧化途径即肌糖原合成也同样有抑制作用。FFA对葡萄糖的抑制作用呈时间依赖性和浓度依赖性,FFA诱导的葡萄糖氧化抑制发生较早,在脂肪输注1~2h后即可看到;而对非氧化途径的抑制则要4小时以后才能出现。FFA在抑制外周葡萄糖利用的同时,还可刺激肝脏糖异生。高FFA状态下,脂肪酸氧化代谢增强,糖异生底物充足,糖异生反应活跃。过多的脂肪酸还通过影响PKC诱导的IRS-1磷酸化而干扰胰岛素的信号传导。
二:TNF-α
在肥胖者血中,TNF-α升高。TNF-α诱发和加重IR的机制包括直接作用和间接作用。其直接作用是:①TNF-α直接作用于培养中细胞的胰岛素信号传导系统,使GLUT4的表达减少;②TNF-α增强IRS-1和IRS-2的丝氨酸磷酸化,这些底物的丝氨酸磷酸化可引发胰岛素受体酪氨酸自身磷酸化的减少及受体酪氨酸激酶活力的降低。我们观察到TNF-α抑制红细胞膜胰岛素受体的自身磷酸化;③TNF-α显著降低IRS蛋白与胰岛素受体相接的能力以及与下游转导途径(如PI3-K和葡萄糖转运)的相互作用。其间接作用有:①TNF-α刺激脂肪细胞分泌瘦素,后者可引起IR;②TNF-α刺激脂肪分解,提高FFA水平,后者是引起IR的重要代谢因素;③TNFα下调过氧化物酶增殖体(PPARγ)基因的表达,抑制PPARγ的合成和功能;④在IR状态下,TNF-α可抑制脂联素的启动子活性,降低脂联素的表达。
三:瘦素
在肥胖患者,血浆瘦素升高,并与FPG和体脂百分率密切相关,被认为是肥胖和IR的一个标志。瘦素的代谢效应与胰岛素的作用相拮抗,瘦素促进脂肪分解,抑制脂肪合成,刺激糖原异生。它调节糖和脂代谢的作用,独立于其抑制食欲和降低体重的作用。相当于肥胖者血瘦素水平的瘦素浓度可使IRS-1酪氨酸磷酸化减弱,并使Grb2与IRS-1的结合能力降低,影响胰岛素的信号传导。
四:抵抗素
也是脂肪组织分泌的,其基因特异表达于白色脂肪组织。在遗传性和饮食诱导的肥胖小鼠,血清抵抗素显著升高,它也是联系肥胖、IR和糖尿病的重要信号分子,而且下调抵抗素的表达是噻唑烷二酮类药物(TZD)发挥抗糖尿病效应的重要机制。但抵抗素在IR和2型糖尿病发病中的确切地位还有待进一步阐明。
五:脂联素
在动物模型和人体中,均已证实低脂联素血症与IR存在相关性。在脂肪萎缩的IR模型鼠中,联合应用生理浓度的脂联素和瘦素可完全逆转IR,单用两者之一仅部分改善IR。研究表明在肥胖和脂肪萎缩鼠模型中,脂联素降低均参与了IR的发生和发展。提示补充脂联素可能为IR 和2型糖尿病的治疗提供全新的手段。TZD可拮抗TNF-α对脂联素启动子的抑制效应,增加脂联素的表达,改善IR。
六:visfatin
是新近发现的脂肪细胞因子,又称为前B细胞集落促进因子(PBEF),分子量为52kD,在骨髓、肝脏和骨骼肌均有表达,在脂肪细胞系3T3-L1的分化过程中,PBEF的基因表达和蛋白合成均增加。人血浆PBEF水平与腹部脂肪体积呈正相关。在2型糖尿病 KKAy小鼠和高脂饮食的c57BL/6J小鼠也发现血浆PBEF水平与内脏脂肪PBEF 的mRNA水平呈正相关。这些结果提示内脏脂肪分泌大量的PBEF,因此研究者又将其命名为visfatin。整体实验证实visfatin有类似于胰岛素的降血糖作用。visfatin还可激活胰岛素受体及其下游信号分子的磷酸化,但其作用方式不同于胰岛素。visfatin与胰岛素两者间存在差异。研究发现小鼠血浆visfatin显著低于胰岛素水平,空腹时血浆visfatin只有血浆胰岛素水平的10%,在饱腹时只有3%左右。此外其血浆水平的变化受饥饿或进食的影响较小,但前炎症因子TNF-α和IL-6都诱导visfatin的基因表达。内脏脂肪素与IR的关系尚不清楚。
其他因素
引起IR的其他原因还有很多。Lautt假设在肝中存在一种外周胰岛素敏感性的调节系统。餐后高血糖兴奋副交感神经,后者促使肝脏中的胰岛素致敏物质(hepatic insulin-sensitizing substance,HISS)释放。HISS激活骨骼肌对葡萄糖的摄取。在2型糖尿病、肝脏疾病和肥胖等疾病时,存在由于HISS调节障碍所致的IR。研究发现性激素结合蛋白(SHBG)可能也与IR有关。近年来的研究认为肾素血管紧张素(RAA)系统也与IR有关。血管紧张素-Ⅱ(AT-2)是RAA的重要效应分子,可能通过影响胰岛素信号通路、抑制脂肪形成、降低组织血流、促进氧化应激和激活交感神经系统等促进IR的发生。临床研究已显示,阻断RAA能改善胰岛素的敏感性,降低新发糖尿病的发生率,为RAA阻断剂在2型糖尿病和代谢综合征等疾病中的应用提供了依据。
多种因素引起β细胞受损
遗传因素
2型糖尿病的直系亲属和双胞胎糖尿病患者的另1位无糖尿病同胞也存在胰岛素分泌功能降低。因此,认为胰岛素分泌功能的降低可能与遗传有关。凡是参与葡萄糖识别、胰岛素加工或分泌的特异性蛋白基因突变均会导致β细胞功能紊乱。目前已发现少数这类信号蛋白的基因突变,包括葡萄糖激酶、线粒体DNA、胰岛素及参与胰岛素加工的酶等。还有一些可能与β细胞功能缺陷有关的基因如GLUT2、β细胞表面的钾通道蛋白和胰淀粉样蛋白(胰淀素)。
早期营养不良影响胰腺发育而导致胰岛细胞数目减少。胎儿、新生儿及婴儿期低体重是早期营养不良的反映,其后果是:影响胰腺发育而导致胰岛细胞数目减少,在长期胰岛素抵抗重压下易发生β细胞功能衰竭。
高糖-高脂-胰淀粉样多肽毒性
高糖、高脂和胰淀粉样多肽毒性是胰岛β细胞功能受损的重要因素:①高血糖损伤胰岛:在胰岛β细胞,糖的氧化代谢将产生氧自由基,在正常情况下,这些物质能被过氧化氢酶和超氧化物歧化酶代谢。在高血糖状态下,β细胞产生大量的氧自由基使β细胞的线粒体受损。②脂毒性损伤胰岛:脂毒性主要可能通过下列机制影响胰岛功能。FFA浓度增加使胰岛素分泌增加,但在24小时后则抑制胰岛素的分泌;脂肪酸能增加UCP-2的表达,其结果是导致ATP形成减少,降低胰岛素的分泌;脂肪酸和TG诱导神经酰胺合成而导致胰岛β细胞的凋亡。③胰淀粉样多肽(IAPP):近90%的胰岛内有淀粉样变,β细胞减少,胰岛淀粉样变性是2型糖尿病的特征性病理改变。IAPP致β细胞受损的机制可能是淀粉样纤维在β细胞和毛细血管间沉积,嵌入细胞膜,损害了细胞膜对葡萄糖的感知和胰岛素的分泌。
β细胞的数量是决定胰岛素分泌量的关键因素。研究显示,长期慢性高血糖下调胰岛β细胞上葡萄糖激酶的表达,使葡萄糖激酶与线粒体的相互作用减少,诱导β细胞凋亡。不过,β细胞数量减少80%~90%时,才足以导致胰岛素缺乏和糖尿病。因此,在2型糖尿病中,除β细胞数目减少外,还存在其他因素损害了胰岛素的分泌。
胰高血糖素样肽-1缺乏
GLP-1由小肠合成和分泌,在维持胰岛β细胞的葡萄糖敏感性等方面起着重要作用,它通过与β细胞上特异性受体结合,调控细胞内cAMP及钙离子水平,最终起到了强化葡萄糖诱导的胰岛素分泌作用。2型糖尿病患者,葡萄糖负荷后GLP-1的释放曲线低于正常人。
胰岛受损的特征
胰岛素分泌不足
2型糖尿病患者存在空腹和葡萄糖负荷后胰岛素分泌量的不足:①2型糖尿病患者存在高FPG,对β细胞造成持续性刺激,导致基础胰岛素分泌增加。FPG和空腹胰岛素间的关系呈倒“U”形或马蹄形曲线。当FPG从4.4mmol/L增至7.8mmol/L时,空腹胰岛素水平逐步增加,达到对照组的2~2.5倍,这是β细胞对葡萄糖稳态被破坏后作出的适应性(代偿性)反应。当FPG超过7.8mmol/L时,β细胞不再能维持高胰岛素分泌率,而致空腹胰岛素逐渐降低;②在正常人,FPG 4.4mmol/L时,葡萄糖负荷2小时后平均胰岛素浓度为50mU/L,进展至IGT(FPG 6.7mmol/L)时,葡萄糖负荷2小时后胰岛素分泌较上述正常人增加约2倍。只要β细胞能保持这种高分泌率,则可维持糖耐量正常或仅轻度异常。当FPG>6.7mmol/L时,葡萄糖负荷后β细胞不再能维持其高分泌率,胰岛素分泌进行性减少,血糖进一步升高。当FPG达8.3~8.9mmol/L时,葡萄糖负荷后胰岛素的分泌量与正常非糖尿病个体相似,但这种胰岛素分泌量相对于高血糖而言,胰岛素分泌是明显不足的。若FPG进一步升高(>8.3~8.9mmol/L),胰岛素分泌反应逐渐降低。当FPG>11.1mmol/L时,血浆胰岛素对糖负荷的反应明显迟钝。
1相胰岛素分泌缺陷
正常人胰岛素第1相分泌峰值在静脉注射葡萄糖后2~4分钟出现,6~10分钟消失。第1相胰岛素分泌在抑制基础状态下肝糖输出有重要意义。在2型糖尿病早期,第1相胰岛素分泌延迟或消失。在IGT和血糖正常的2型糖尿病一级亲属中也可观察到胰岛素第1相分泌缺陷,故认为这种缺陷可能不是继发于高血糖的毒性,而是原发性损害。早期胰岛素分泌有重要生理意义,可抑制肝葡萄糖输出,抑制脂肪分解,限制FFA进入肝脏,减轻负荷后高血糖的程度,使血糖曲线下降,并减轻负荷后期的高胰岛素血症。正常人OGTT或馒头餐时,血浆胰岛素分别约于30 或60分钟达峰值,此为负荷后早期胰岛素分泌。2型糖尿病患者OGTT 30分钟时,血浆胰岛素明显低于正常人,相对于其有显著增高的血糖而言,早期胰岛素分泌严重不足。评估早期胰岛素分泌的一种实用方法为OGTT中30分钟胰岛素与基线值差别及葡萄糖与基线值差别两者的比值。早期胰岛素分泌障碍的后果为糖负荷后显著高血糖,刺激胰岛素分泌,使胰岛素往往于2小时达峰值。同时可使餐后血非酯化脂肪酸得不到有效地控制,并出现餐后高TG血症。
胰岛素分泌脉冲紊乱
正常人在空腹时,胰岛素的脉冲分泌周期约为13分钟。胰岛素脉冲分泌有助于防止靶组织中胰岛素受体水平的下调,维持胰岛素的敏感性。反之,持续的高胰岛素血症将导致胰岛素受体水平下调,引发IR。在2型糖尿病中,胰岛素分泌正常的13分钟间隔脉冲消失,出现高频率(5~10分钟)脉冲,为2型糖尿病的早期标志。在2型糖尿病一级亲属中可观察到正常的胰岛素分泌脉冲消失,提示胰岛素分泌脉冲异常可能是原发性损害。
胰岛素原分泌增多
胰岛素原的生物活性只有胰岛素的15%。胰岛素原在高尔基体激素原转换酶2(PC2)、激素原转换酶3(PC3)和CPH的作用下转变为胰岛素,同时产生C肽和去二肽胰岛素原。高血糖刺激胰岛素原和PC3的合成,而PC2和CPH不受血糖的影响。在2型糖尿病中,胰岛素原与胰岛素的比值增加,不利于血糖的控制。
2型糖尿病发病涉及胰岛素作用和胰岛素分泌两个方面的缺陷,二者与遗传因素和环境因素均有关,环境因素通过遗传因素起作用。糖尿病遗传易感个体的早期即存在胰岛素抵抗,在漫长的生活过程中,由于不利环境因素的影响或疾病本身的演进,胰岛素抵抗逐渐加重。为弥补胰岛素作用的日益减退及防止血糖升高,β细胞的胰岛素呈代偿性分泌增多(高胰岛素血症)。在此过程中,β细胞增生和凋亡均增加,但后者更甚。当β细胞分泌能力不足以代偿胰岛素抵抗时,即出现糖代谢紊乱;首先是餐后血糖升高(IGT期)。当胰岛素抵抗进一步加重,β细胞因长期代偿过度而衰竭时,血糖进一步升高,终致糖尿病。高血糖又可抑制葡萄糖介导的β细胞胰岛素分泌反应,增强胰岛素抵抗(葡萄糖毒性,glucose toxicity),并形成胰岛素分泌与作用缺陷间的恶性循环。
