
糖代谢对神经系统非常重要,神经元能量的获得依赖葡萄糖。糖尿病时出现的细胞代谢异常,不可避免地也会影响神经系统。因此,糖尿病神经病变是糖尿病很常见的合并症,有人统计糖尿病性神经病变的发生率可达10%~50%。近年通过对动物和人糖尿病时细胞代谢异常的研究,发现糖尿病时周围神经中有肌醇(myo-inositol)的减少及山梨醇的积存。肌醇来源于食物及肾脏合成。因细胞的钠及能量依赖摄取系统的作用,神经细胞中肌醇的浓度比血清中约高100倍。高血糖及高山梨醇水平均抑制此种摄取,因而减低肌醇水平。肌醇为多磷酸肌醇的前体,后者为神经细胞膜的组成成分。肌醇的减少会影响细胞膜的功能,改变Na+/K+三磷腺苷酶的活性,从而影响兴奋细胞的传导性。
另外,高血糖时神经元的山梨醇通路活性增加,山梨醇和果糖积存。而这些物质通透性不好,因此渗透压发生变化,使神经内膜液体增加,施万细胞皱缩,对神经产生一定影响。也有人发现糖尿病患者有脂质代谢异常,导致神经的脂质合成异常和组成髓鞘的脂质比例异常,但尚未得到一致的认识。此外,糖尿病患者还有蛋白质代谢的紊乱。近年有人发现糖尿病动物的神经轴索早期即有逆行传送障碍。除了代谢机制外,糖尿病患者有大血管和微血管的改变。大血管的改变可能是通过促进动脉粥样硬化造成,因而促使脑缺血性疾病的发生率增加。微小血管的改变,如累及供养神经的微血管,则可发生单神经病变或多发性神经病变。近来对微血管病变在多发性神经病变中所起的作用由于得到实验室的证据而更加以强调。因此,很可能代谢机制和血管机制都是发生糖尿病神经系统合并症的重要原因。
此外,还发现有蛋白酶的不足,蛋白代谢的紊乱。但何者为引起糖尿病性神经病变的主要机制尚未得到一致的认识。既往对肌醇的研究主要依据糖尿病动物。近年Dyck等对临床有神经病变的糖尿病患者、无神经病变的糖尿病患者以及对照组的神经活检结果进行研究,发现糖尿病患者神经内膜的肌醇并未减少。山梨醇沉积的问题,十余年来应用山梨醇还原酶抑制剂治疗糖尿病神经病变并未取得显著成功,可能能延缓末梢性对称性多发性神经病变的进展,但似未能逆转。近来则有不少证据提示神经的血供不足是引起神经病变的重要发病因素。通过改善供血可以使神经病变有所好转;如与山梨醇还原酶抑制剂(ARI)合用,则可进一步提高疗效。另外还发现多元醇通道的改变,如多元醇通道的活跃,使烟酰胺腺嘌呤磷酸二核苷酸磷酸(NADP)减少,这影响了谷胱甘肽氧化还原功能,从而影响细胞对氧自由基的自我保护作用,导致血管内皮细胞受到损害。氧自由基也会中和氧化氮。因而削弱氧化氮介导的神经营养血管的扩张作用。也有人发现ARI的作用与氧化氮介导的血管松弛作用有关。如糖尿病鼠予以氧化氮合成酶抑制剂,可以完全阻断ARI改善神经传导速度(NCV)和血液循环的作用。
糖尿病常伴有大血管及微血管的改变。大血管的改变可能是通过血管内膜的损伤,促进动脉粥样硬化造成,微小血管病变可能与糖化作用(glycosylation)产物(晚期糖基化终末产物;advanced glycation end product;AGE)及自由基等有关。微血管病变是发生单神经病变及多发性神经病变的基础,已有不少实验室及病理学的证据。近年也有人发现多灶性糖尿病性神经病变有血管炎性改变。因此,很可能代谢机制和血管机制都是发生神经系统合并症的重要原因。尤其近年有不少学者明确提出:血管内膜的损伤,导致神经微循环的障碍,是发生糖尿病神经病变的重要机制。由此也可能因而改变我们对糖尿病神经病变治疗的策略。
高血糖促使内皮细胞及大动脉等的甘油三酯增加,激活蛋白激酶C等,导致花生四烯酸释放及前列腺素E2生成增加。而这些对钠、钾离子及ATP酶均有抑制作用,导致神经传导速度减慢。
遗传因素:遗传因素在糖尿病神经病变的发生中可能起一定作用,即“易感性”。有的控制并不好的严重糖尿病患者,多年不出现神经病变。而有的轻症糖尿病患者却出现神经病变。提示可能有遗传因素在起作用。
总的看来糖尿病性神经病变不是单一发病机制引起,而是遗传因素,神经缺氧/缺血,氧化应激,多元醇旁路过度活跃,晚期糖基化终末产物增加,γ-亚麻酸缺乏,蛋白激酶C增加,生长因子缺乏,及免疫异常等因素综合引起(下图)。
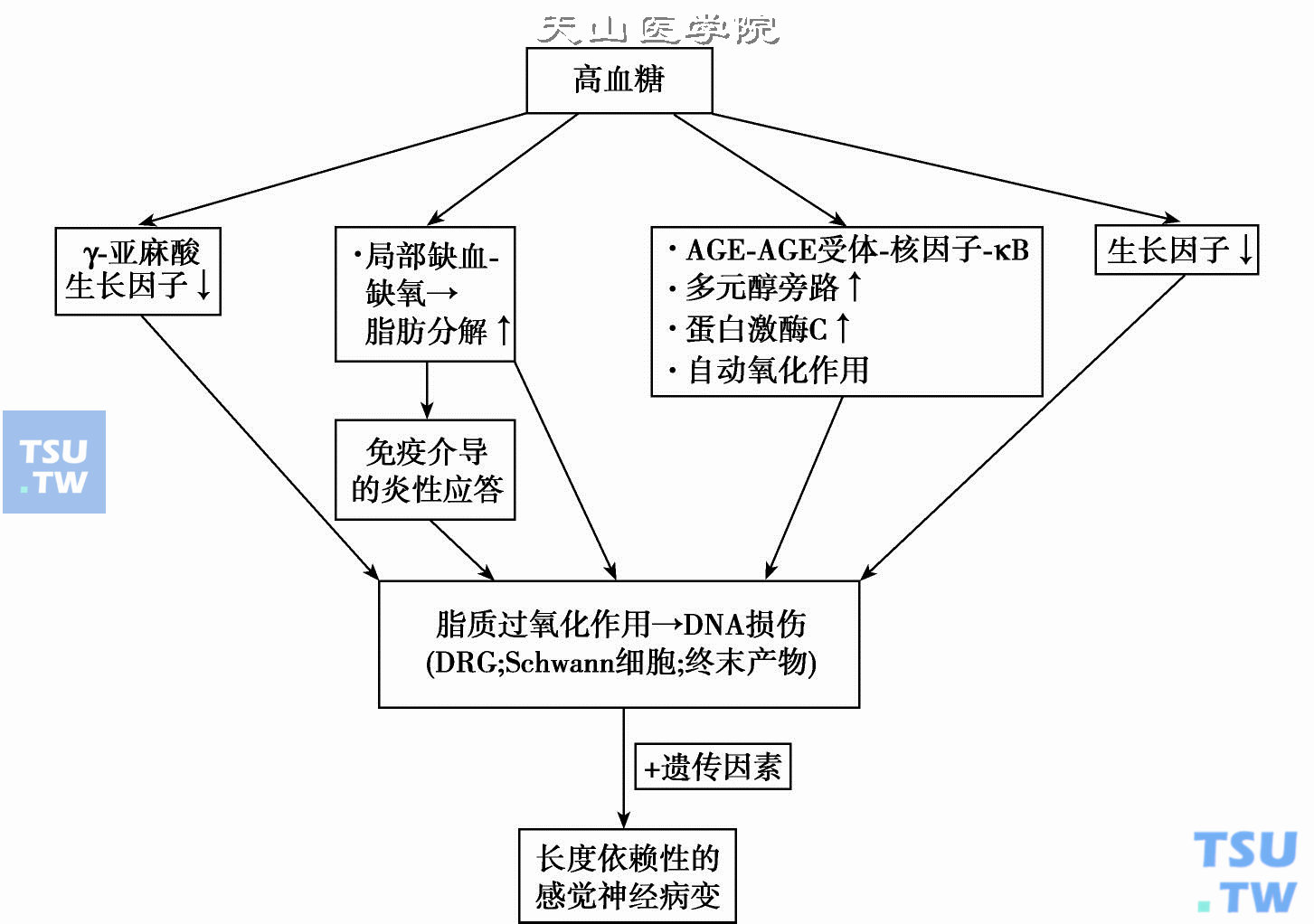
糖尿病性神经病变发病机制示意图;AGE,晚期糖基化终末产物;DRG,背侧根神经节(引自:Joslin’s Diabetes Mellitus 14th ed. 2006,中文版2007 p. 881)
