
硬皮病是以局限性或弥漫性皮肤及内脏器官结缔组织的纤维化或硬化,最后发生萎缩为特点的疾病。硬皮病的分类:
一、局限性硬皮病(localized scleroderma)
1.硬斑病(morphea)
- 点滴状硬斑病(guttate morphea)
- 斑块状硬斑病(plaque-like morphea)
- 线状硬皮病(linear scleroderma)
- 泛发性硬斑病(generalized morphea)
2.深部硬斑病(morphea profunda)
3.致残性全硬化性硬斑病(disabling pansclerotic morphea)
4.特殊类型硬皮病
- 大疱性硬斑病(bullors morphea)
- 结节性硬皮病(nodular或keloidal scleroderma)
二、系统性硬皮病(systemic scleroderma)也称进行性系统性硬化症(progressive systemic sclerosis,PSS)。
- 肢端硬皮病(acroscleroderma)也称肢端硬化病(acrosclerosis)。
- 弥漫性硬皮病(diffuse scleroderma)。
- CREST综合征(calcinosis cutis,Raynaud's phenomenon,esophageal dysfunction,sclerodactyly,telangiectasia)。
两类硬皮病有不同的临床表现,它们的内脏损害和预后不同。局限性硬皮病病变主要局限于皮肤,早期的损害往往只限于肢体远端,或手指以及面部的皮肤,内脏一般不受累,预后较好;系统性硬皮病则有广泛分布的皮肤硬化,雷诺现象和多系统受累,预后不定,大多较好,弥漫性者预后不良。在硬皮病的局限性和弥漫性两极型之间可见一些中间型,如局限性硬皮病中的泛发性硬斑病和系统性硬皮病中的肢端硬皮病和CREST综合征,因此本病亦为病谱性疾病。
两类硬皮病均以女性发病率较高,与男性患者相比为3:1。发病年龄:局限性者多数在11~40岁,系统性者在21~50岁。
病因及发病机制
病因不明,关于硬皮病的发病学说有:
一、免疫学说
本病常合并LE、皮肌炎、类风湿关节炎等自身免疫性疾病,血清中存在多种自身抗体,虽然这些自身抗体在发病中的作用尚不清楚,但已发现与疾病的型别相关,如抗着丝点抗体对CREST综合征有高度特异性,抗Scl70抗体与PSS相关,并常标志着病人有肺部受累和病情较重。直接免疫荧光发现,在PSS病人的皮肤、肌肉和肾脏血管的平滑肌和弹力层中有IgM、IgA、IgG的沉积。病人中还有高γ球蛋白血症、循环免疫复合物增加等,均提示本病属自身免疫性疾病范畴。
在细胞免疫方面,已经证明硬皮病早期损害的真皮和皮下组织中有单一核细胞浸润,这些浸润细胞多为T淋巴细胞,并几乎都表达Ia抗原,说明这些T淋巴细胞处于活化状态。在人类胚胎的组织培养中,来自PSS病人的淋巴细胞能破坏培养的胚胎成纤维细胞。在体外用植物血凝素(PHA)刺激患者的淋巴细胞,可引起淋巴因子释放,能刺激成纤维细胞产生胶原。骨髓移植后发生的慢性移植物抗宿主病,其皮损在临床和组织学上均与硬皮病相似,进一步支持细胞免疫在硬皮病发病机制中的作用。
二、胶原合成异常学说
现在已经明确,硬皮病皮肤的绷紧和坚硬度是由于新合成的胶原替代了大部分或全部的真皮和(或)皮下脂肪,使皮肤与其下组织紧贴。其早期组织学表现为:胶原束肿胀,整个真皮增厚,成纤维细胞增多。这与成纤维细胞合成胶原增加有关。有研究发现,硬皮病患者皮肤成纤维细胞在培养中比正常成纤维细胞合成更多胶原,这种差别在体外培养中可传15代之多。患者真皮下部和皮下组织的成纤维细胞活性较真皮上部的更强,而正常皮肤中,真皮上下部的成纤维细胞之间没有功能差别。真皮和皮下成纤维细胞胶原产生增加的刺激因素不明,有报道前胶原氨基末端伸长的肽即前胶原肽(proeollagen peptides)通过反馈抑制能调节正常成纤维细胞的胶原合成,将前胶原肽加入硬皮病成纤维细胞培养基中,其胶原合成过度可基本恢复正常,而不影响其他蛋白质的形成。在硬皮病中,由于这些前胶原肽从胶原上延迟切除,减弱了其反馈抑制作用,从而使胶原纤维形成增加。最近的观察提示致敏淋巴细胞也可能在其中起作用。
三、血管学说
雷诺现象常为PSS早期表现,PSS中可见到的毛细血管扩张发生部位通常是雷诺现象的好发部位,即面部、舌、唇、手和上胸。在硬皮病中,95%的患者具有雷诺现象,其中75%以雷诺现象为首发症状,这些病人往往具有内脏损害,且硬皮病的预后和最终结局很大程度上取决于血管损害的范围和严重性。
其血管变化主要包括:血管舒缩功能障碍,小血管结构异常,动脉内膜增殖,微循环闭塞,血小板活性增强,红细胞变形能力下降和血栓形成。主要影响皮肤、肺、冠状血管、指(趾)和肾叶间动脉。这些变化常出现在疾病早期,组织硬化之前,提示血管变化是发病的基础环节。认为硬皮病原发损害发生在血管内。
研究表明,血管内皮细胞的损害和功能失调是其中的中心环节。内皮细胞的损伤可使内皮细胞肿胀,毛细血管扩张,血管通透性增加,血管壁嗜伊红蛋白沉积,血管内膜增殖,以至于弥漫性血管和微血管堵塞,使皮肤、肺、肾等重要脏器处于低灌注和慢性缺血状态,使组织处于缺氧状态。实验表明,在低氧状态时,成纤维细胞寿命和克隆生长延长,促进组织纤维化,从而启动和维持硬皮病的急慢性表现。导致内皮细胞损伤的有以下几种解释:
- 有研究证实部分硬皮病患者血清中存在内皮细胞特异性细胞毒因子,如酸性溶酶体水解酶、白三烯B4(LTB4)和抗血管内皮细胞抗体等;
- 由于细胞免疫反应,病人的Th细胞和单核细胞处于活化状态,血清中IL-1、IL-2、TNF、sIL-2R和可溶性CD4抗原水平增高,这些可溶性介质可以直接或间接抑制内皮细胞生长,损伤其超微结构;
- 也有证据表明,震动能与内皮细胞损害有关,使用震动性工具者可发生雷诺现象,并可引起指(趾)硬皮病;④病人血小板的激活可能继发于内皮细胞损伤和活化,它与内膜增殖性损害有关。
总之,硬皮病的发病机制尚不清楚,它的发病过程有上述多种因素的参与。有作者对其发病机制提出如下一元性假说:全身性血管收缩引起血管痉挛,后者导致肺动脉高压,心、肾病变。加上局部因素,引起雷诺现象,反复和长期的血管痉挛发作,导致组织贫血和血管内皮细胞损伤,其后修复,致血管内膜增生。免疫系统异常情况下,组织贫血和坏死有利于发生细胞免疫,因此致敏淋巴细胞能刺激成纤维细胞,产生皮肤和肺的纤维化。血管收缩发生的原因不明,可能由于血管和平滑肌上的受体缺陷,引起血管平滑肌神经支配异常。
症状表现
局限性硬皮病
1.点滴状硬斑病 多发生于上胸、颈、肩、臀或股部。损害为黄豆至五分硬币大小,白色或象牙色的集簇性或线状排列的斑点,圆形,有时稍有凹陷。病变活动时,周围有紫红色晕。早期质地硬,后期质地可变软或有“羊皮纸”感觉。
病变发展很慢,向四周扩展而相互融合或持续不变。某些皮损可消退,局部残留轻度萎缩的色素沉着。
2.斑块状硬斑病 较常见。最常发生于腹、背、颈、四肢、面。初呈圆或不规则淡红色或紫红色水肿性斑片,经数周或数月后扩大,直径可达1~10cm或更大,呈淡黄或象牙色。表面干燥平滑,具蜡样光泽,周围有轻度紫红色晕,触之有皮革样硬度,有时伴毛细血管扩张。局部无汗,亦无毛发(图3-12-26)。经过缓慢,数年后硬度减轻,渐渐萎缩,中央色素脱失。可侵及真皮及浅表皮下,但仍可移动。皮损的数目和部位不一,多数患者只有一个或几个损害,有时呈对称性。皮损在头皮时可引起硬化萎缩性斑状脱发。
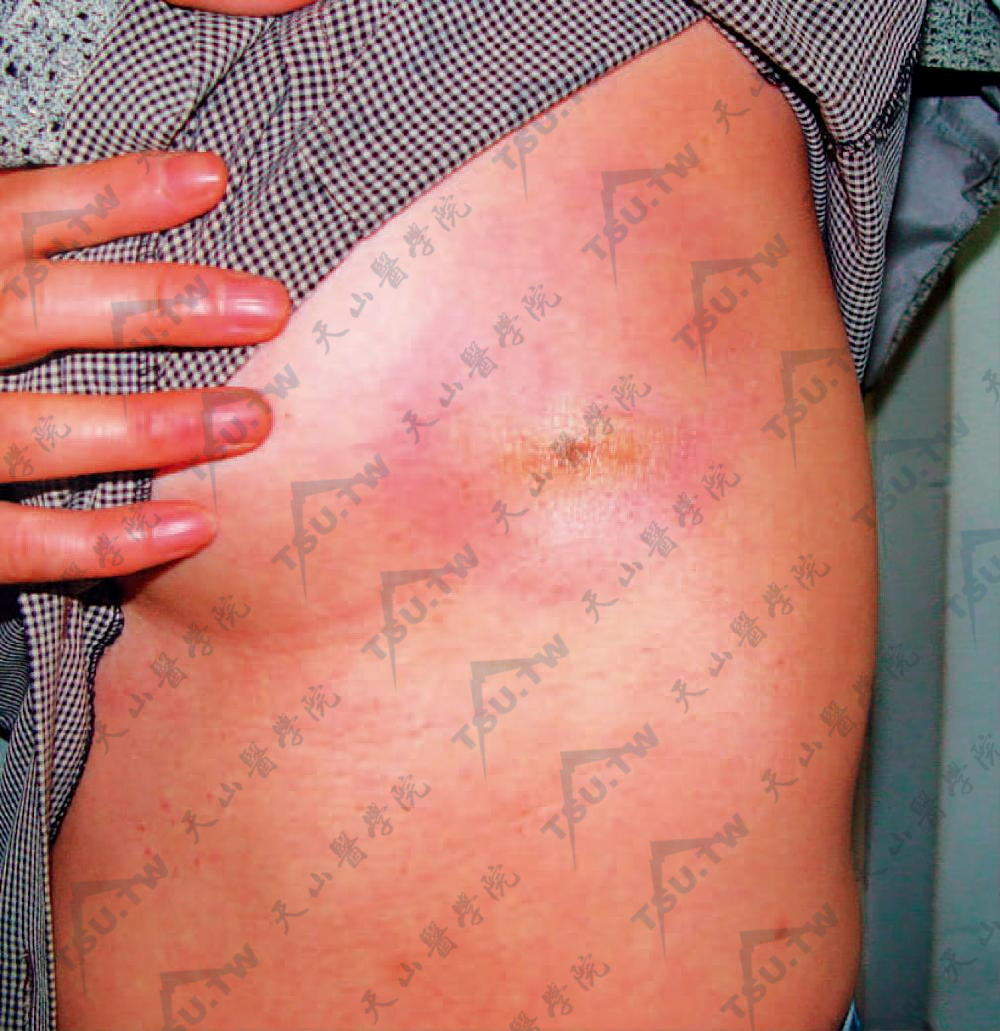
图3-12-26 胸前左侧大片淡红色斑,水肿样,中央淡黄白色,发亮。表皮轻度萎缩,触之硬
3.线状硬斑病 皮肤硬化常沿肋间神经或一侧肢体呈带状分布,亦可发生于前额近正中部向头皮延伸呈刀砍形,局部皮损显著凹陷,常开始即成萎缩性,皮肤菲薄不发硬,程度不等地贴着于骨面上(图3-12-27,图3-12-28)。额部带状硬斑病大多单独出现,某些病例可合并颜面偏侧萎缩。带状损害常累及浅部及深部皮下层如皮下脂肪、肌肉和筋膜,最终硬化固定于下方的组织,常引起严重的畸形。在肘、腕、指等关节面越过时,可使关节活动受限,并发生肢体张弓状挛缩和爪状手。
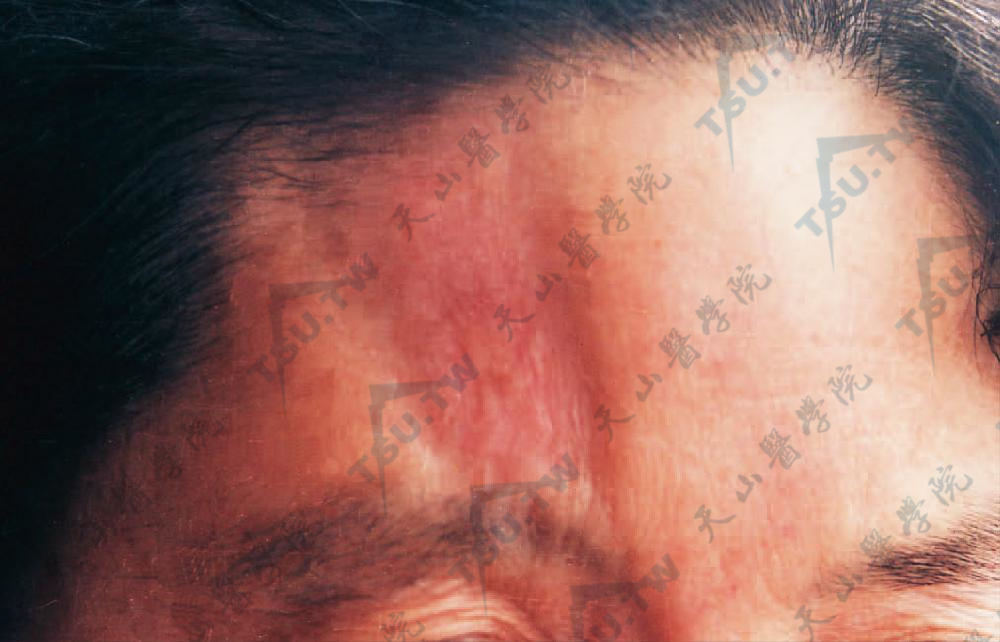
图3-12-27 皮肤硬化于前额近正中部向头皮延伸呈刀砍形,局部皮肤显著凹陷,开始即成萎缩性
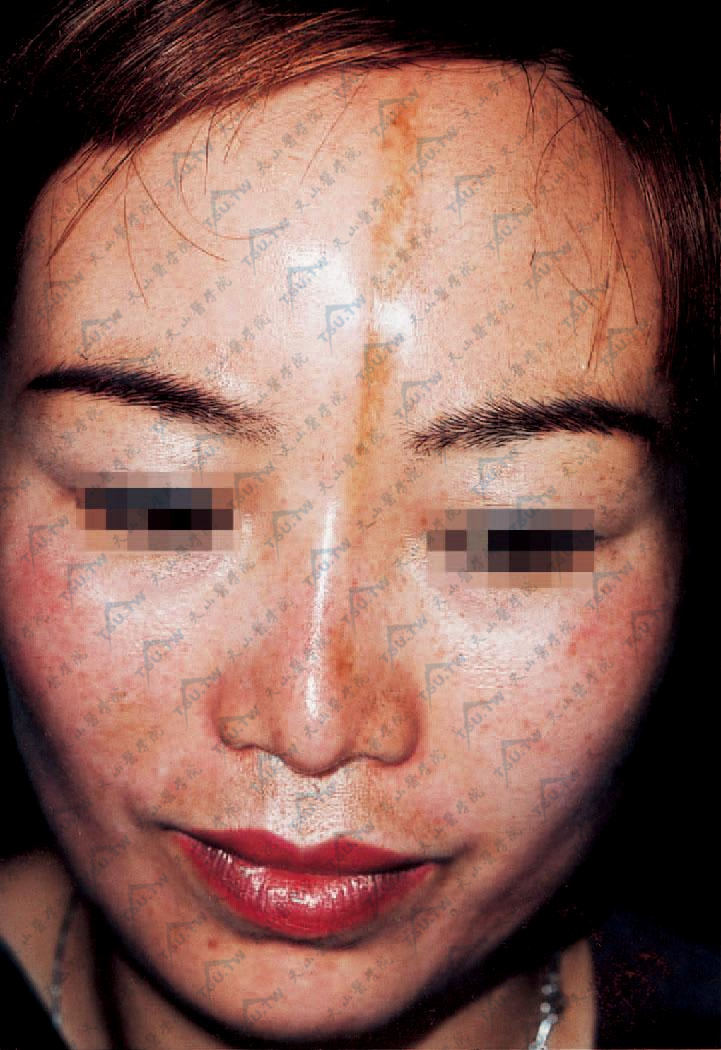
图3-12-28 前额正中稍偏左侧约6cm×0.5cm条状淡褐色萎缩性斑片,上至发际,下至鼻梁中下方,表面光滑、凹陷,与皮下粘连
4.泛发性硬斑病 点滴状、斑块状和线状等类型损害可部分或全部合并存在,损害很多,分布于全身各个部位,但很少累及面部,损害常有融合倾向,常可合并关节痛、腹痛、神经痛、偏头痛和精神障碍,偶可转为系统性硬皮病。
5.深部硬斑病(morphea profunda)Su及Person(1981年)报道的病例,即深部脂膜及筋膜硬化的硬斑病,有时亦侵及真皮、真皮深层及浅部肌肉。
6.致残性全硬化性硬斑病(disabling pansclerotic morphea)是另一种类型硬斑病,发生于儿童,发病年龄1~14岁,多见于女孩。真皮、皮下组织、筋膜、肌肉及骨骼发生炎症和硬化,好发于四肢,特别是伸侧,手、足、肘和膝呈屈曲挛缩,很少侵犯内脏,无雷诺现象,患者可有硬化性苔藓样皮损,身体其他部位可有典型硬斑病表现。
此外,还有学者报道过硬斑病中的一些特殊类型及特殊表现:
大疱性硬斑病(bullous morphen)主要表现是在硬斑基础上出现大疱,常于下肢,提示淋巴管扩张及下肢静脉压力较大是产生水疱的原因之一(图3-12-29,图3-12-30)。
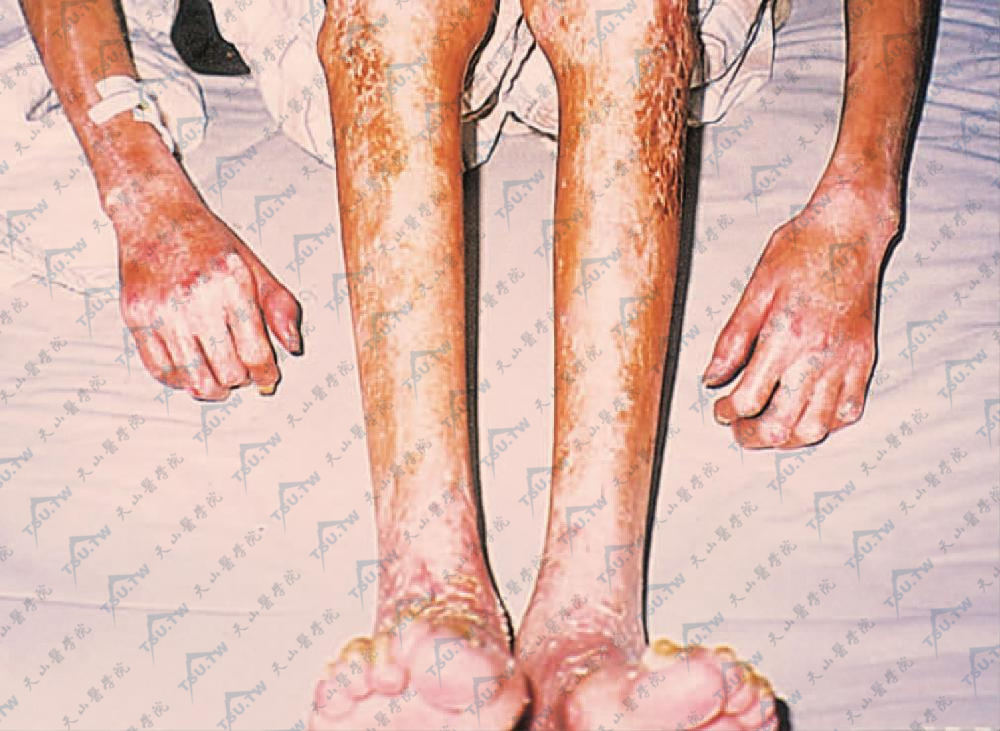
图3-12-29 大疱性硬斑病 四肢远端皮肤萎缩、硬化、关节挛缩,活动明显受限(中国医学科学院、皮肤病研究所 贾 虹提供)
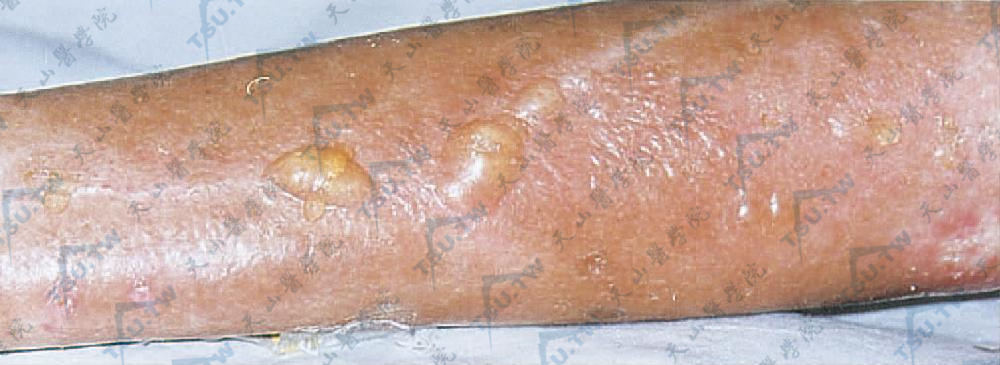
图3-12-30 大疱性硬斑病 左上肢硬化皮肤上沿血管分布的水疱(中国医学科学院、皮肤病研究所 贾 虹提供)
结节性硬皮病(nodular或keloidal scleroderma)少数硬皮病患者在皮肤硬化区中出现瘢痕疙瘩样结节,称为结节性硬皮病(图3-12-31,图3-12-32)。
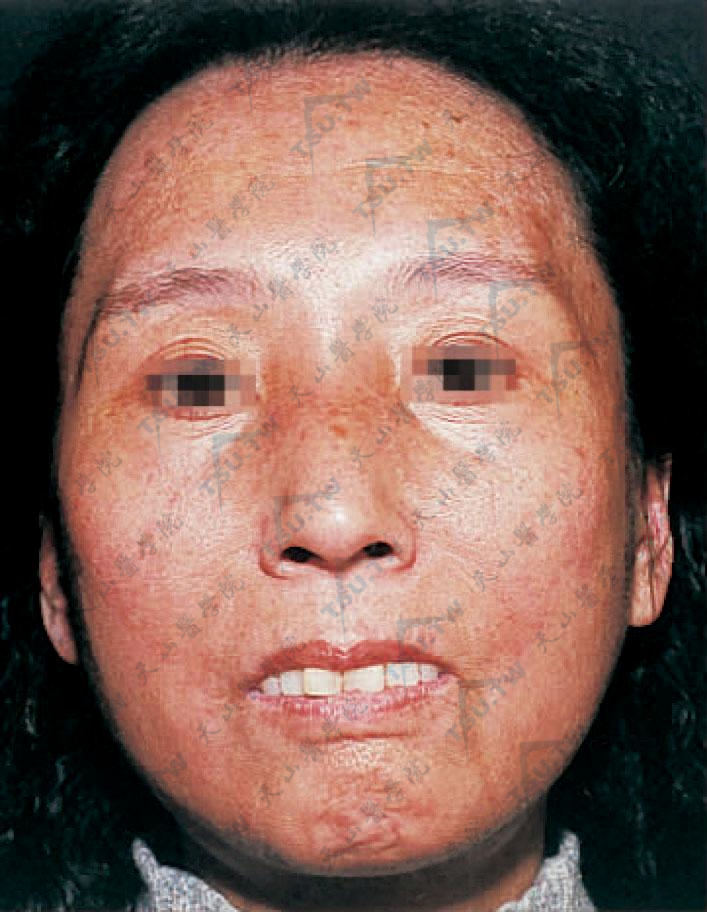
图3-12-31 结节性硬皮病 面部皮肤发亮,缺乏表情呈假面具样(北京协和医院皮肤科 马东来提供)
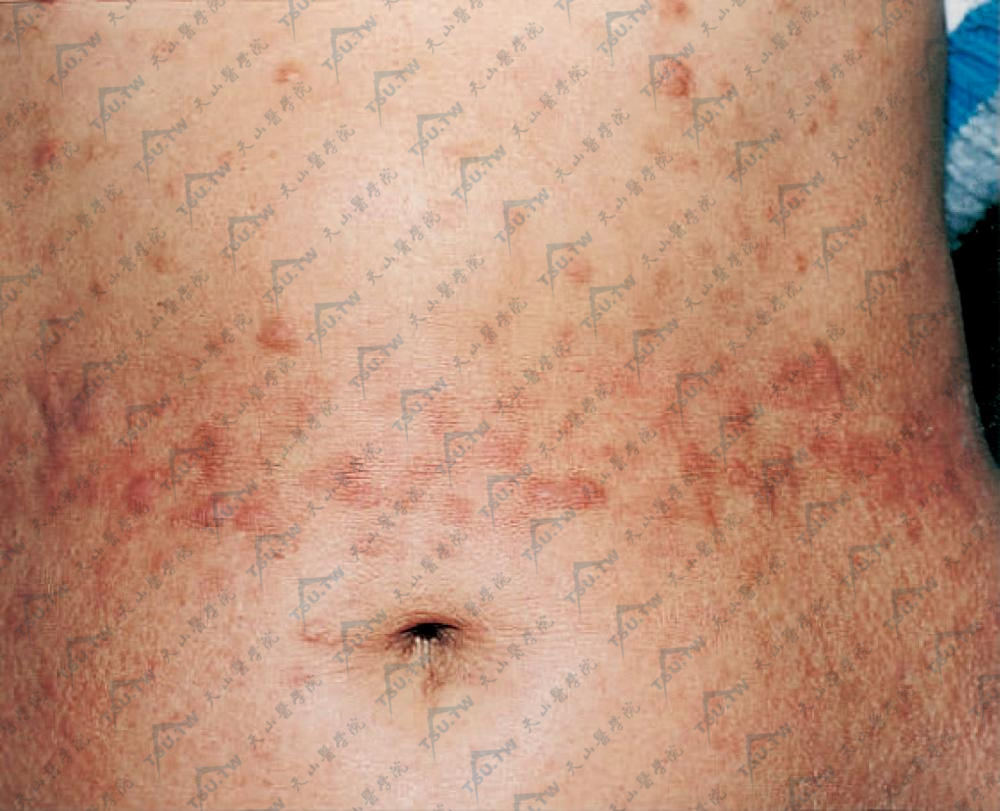
图3-12-32 结节性硬皮病 腰部可见许多绿豆至黄豆大的结节,呈肤色或淡红褐色(北京协和医院皮肤科 马东来提供)
系统性硬皮病
1.前驱症状:多数患者有雷诺现象、关节痛、神经痛、不规则发热、食欲减退、体重下降。
2.皮肤症状:发病常自手、足和面部开始,渐扩展至前臂、躯干上部等处,呈对称性。局部先发生红斑肿胀,压之无凹陷,继之皮肤坚实发亮,灰黄色似蜡样,可有色素异常和毛细血管扩张。皮肤因与皮下组织粘连而用手指不能提起褶皱。面部表情丧失呈假面具样,鼻尖似鹰嘴,口唇变薄且收缩成放射状沟纹,口裂狭小。指关节活动受限可呈爪状手,肘、膝关节可屈曲挛缩。胸部皮肤受累可影响呼吸运动。皮肤、皮下组织、肌肉均可萎缩,甚至皮肤直接贴于骨面。损害处毳毛可脱落,出汗减少,皮脂缺乏。甲可增宽,表面有纹,易碎或变薄,脱落。皮肤表现常见有:
肢端硬皮病 肢端硬化者可表现为典型指(趾)硬皮病(sclerodactyly),即手指逐渐变细,皮肤光亮绷紧,多发生雷诺现象,轻者只有血管痉挛,没有皮肤持久性改变;重者为发作性血管功能不全,引起指尖溃疡或坏疽。约占系统性硬皮病的95%,女性多见。常先有雷诺现象。皮肤硬化白手部开始,继之累及前臂、面、颈、躯干上部和下部,但躯干和下肢皮肤硬化程度较轻。手指部常有皮肤钙沉着及远端指骨吸收。病情进展缓慢,皮肤硬化区可自行缓解。

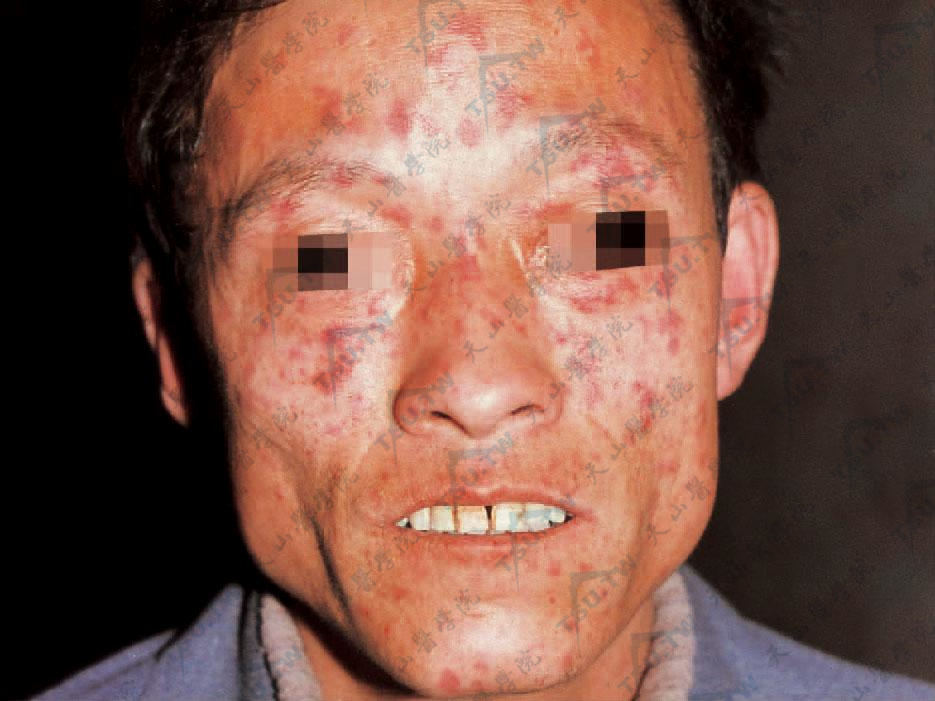
肢端硬皮病面部皮肤坚实发亮,灰黄色似蜡样,呈假面具样。
鼻尖、口唇变薄,口裂狭小。手指变细,皮肤光亮绷紧
弥漫性硬皮病 约占系统性硬皮病的5%。男女发病相近。皮肤硬化常自躯干开始,后累及四肢、面部。萎缩较肢端型轻。无雷诺现象,也无肢端硬化和皮肤钙沉着,罕见远端指骨吸收,病情进展迅速,经2年左右全身皮肤大部均硬化,晚期侵犯内脏,预后差。皮损罕见自动消退者,可迅速死亡。死亡率为肢端型的3倍。
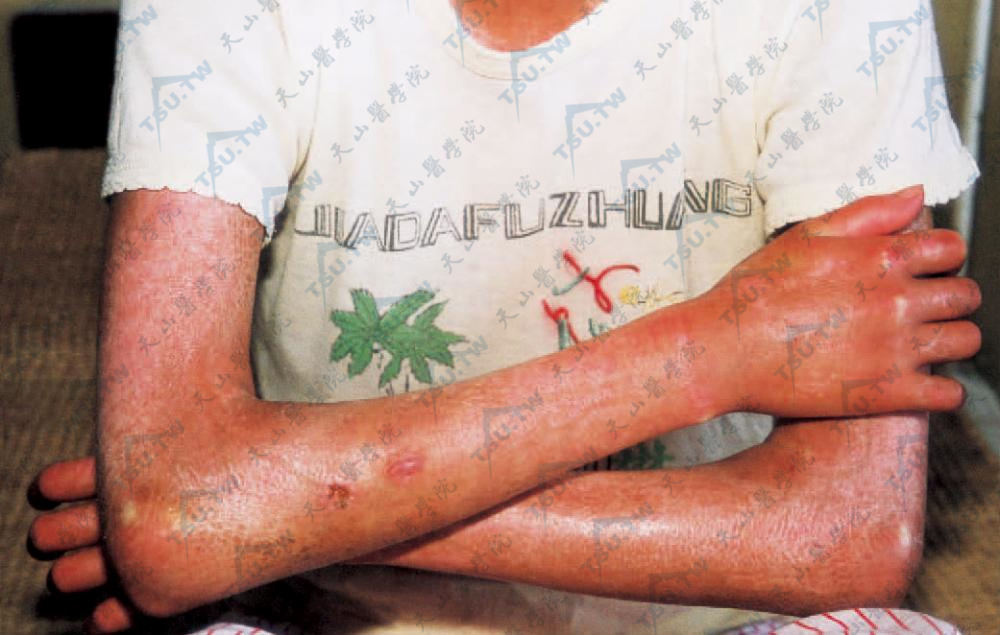
弥漫性硬皮病 四肢、躯干皮肤弥漫性硬化,光亮绷紧。手指弯曲肿胀,有雷诺现象
CREST综合征 1964年Winterbauer将指(趾)硬皮病合并有皮肤钙着、雷诺现象和毛细血管扩张者称为CRST综合征(CRST syndrome),又称蒂比耶日-魏森巴赫综合征(Thibierge-Weissenbach syndrome)。因大多数患者有食管蠕动功能异常,故Rodnan称其为CREST综合征,即除上述四症状,再加食管功能障碍。此为PSS的亚型,病程缓慢,预后良好。
CRST综合征 面部皮肤毛细血管扩张及硬皮病表现
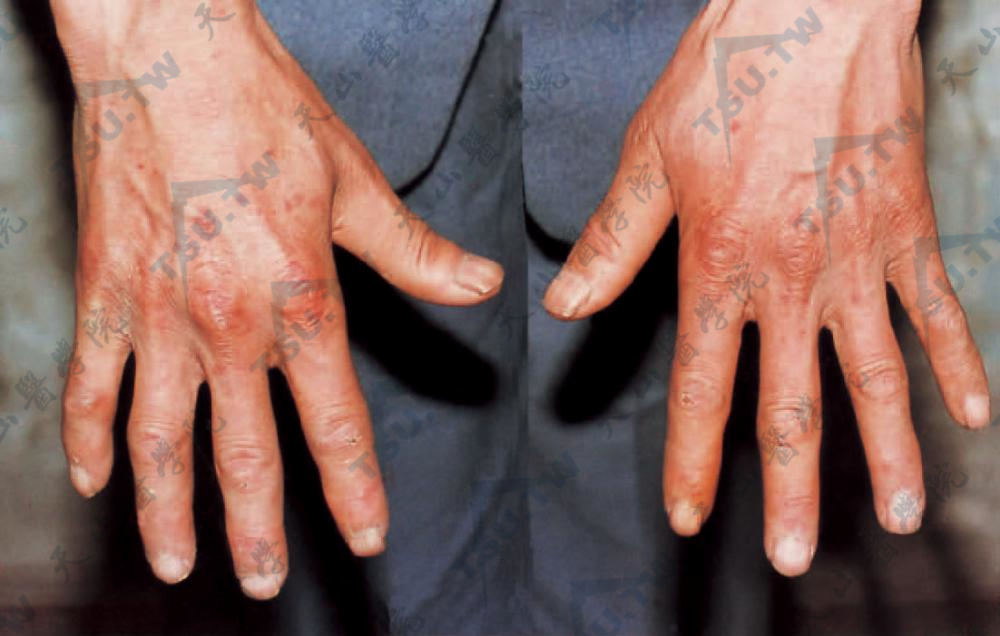
CRST综合征 双手有肢端硬化及毛细血管扩张
硬皮病常伴有色素异常,有以下三种表现:
- 泛发性色素沉着或暴露部位和黏膜色素增加,色素沉着不限于硬化部位,类似于肾上腺皮质功能不全,但肾上腺皮质功能正常;
- 硬化部位局灶性色素沉着或色素脱失;
- 色素全部脱失,色素脱失斑上出现毛囊周围色素沉着斑。常见于上胸和背部,头皮、发线、面和前臂伸侧以及耳廓。
大多数患者甲后皱襞有线状毛细血管扩张。毛细血管扩张几乎无一例外地见于硬皮病,其特点为呈线状、卵圆形、正方形和多角形的境界清楚、局限性毛细血管扩张性斑,粉红至鲜红色,1~5mm大小,少数可达1~2cm。常见于面部、手、唇和颊黏膜。仔细检查有些损害中可见纤细的毛细血管扩张性血管,另一些损害仅能见均匀的红斑,损害境界清楚或稍模糊。
皮肤钙沉着一般见于硬皮病病程晚期,主要在大关节周围和手指,不影响功能,但常发生疼痛。
3.黏膜损害 舌、齿龈、软腭、咽喉、阴道黏膜等均可硬化萎缩。舌肌可萎缩,舌系带硬化缩短,致舌不能伸出口外。硬化可累及腭垂、腭帆、眼球及睑结膜的结缔组织和肌肉而引起咽门窄小、眼闭合不全、眼球转动受限等表现。不少患者可有干燥综合征的表现,如口腔和喉干燥,唾液腺功能减退等。
4.系统病变 系统性硬皮病可侵犯内脏各器官,以关节、肺、食管多见,其他如心、肠道、胃、肾、肌肉、肝、脾、骨髓、淋巴结、中枢神经系统、内分泌腺等。内脏损害也可发生于皮肤症状之前,但5%以下的病人不发生皮肤增厚和其他皮肤表现,称为无皮肤硬化型系统性硬化症(systemic sclerosis sine scleroderma),又称无皮肤损害的内脏硬皮病。
- 骨、关节:大多数患者有关节病变,表现为关节痛和关节炎,可有少量关节渗液。大小关节同时受累,而以手部小关节常见,可有畸形。Ⅹ线表现关节间隙狭窄和关节面的硬化。骨变化以指骨的吸收较特殊,临床可见指骨变短变细,Ⅹ线变化常为骨疏松,也可发生骨硬化、骨破坏、骨变形、骨萎缩。牙槽突骨可萎缩,牙周间隙增宽,牙齿脱落显著。下颌关节Ⅹ线检查亦有关节间隙变窄,可有下颌角的骨吸收,此与面部皮肤的拉紧、咬肌和翼状肌的萎缩及口裂变小有关。
- 食管:食管远端运动障碍是内脏受累的最常见症状,约42%的患者有食管受累的临床表现,主要症状为有不同程度的吞咽困难,进固体食物时尤为明显。有时累及食管下端的括约肌,导致胃内容反流。669%的患者有食管Ⅹ线异常改变。钡剂透视见整个食管扩张和食管下2/3蠕动减弱以至消失,而食管下1/3处常见有狭窄。
- 胃肠道:胃的变化较少,常侵犯十二指肠和小肠。表现为腹痛,腹泻与便秘交替,有类似麻痹性肠梗阻的表现及吸收障碍综合征。Ⅹ线检查示消化道扩张,钡剂排空和通过时间延长。结肠可见憩室,但缺乏临床症状。
- 肺:约25%的患者有肺部弥漫性间质性纤维化。有时伴支气管扩张和肺气肿,少数可见囊肿形成。胸膜炎少见。临床上约半数患者有呼吸困难,而大约95%硬皮病患者肺功能试验异常:肺活量降低,气体弥散障碍,肺顺应性低,提示肺泡弹性减退和肺动脉高压。系统性硬皮病合并肺癌的病例报道近来渐增多,特别是肺泡细胞癌。
- 心脏:心脏病变与心肌纤维化和肺小动脉炎密切相关。有研究表明,大部分患者有轻微的左心功能不全迹象。心肌受累约占10%,可为继发性,也可为原发性心肌损害。同时可侵犯心包及心内膜。临床表现有气急、胸闷、心悸和各种房性与室性心律失常,也可有踝部水肿、呼吸困难,有时可发生心绞痛或晕厥,甚至心力衰竭。Ⅹ线表现心脏扩大、心搏减弱或心包积液。心电图示有心房颤动、期前收缩、低电压、房室传导阻滞、ST段偏移及T波平坦等改变。
- 肾脏:硬皮病的肾脏病变以慢性型居多,表现为患病2~3年后逐渐发生轻度蛋白尿和镜下血尿,常为疾病严重的表示。一些作者在尸解及肾组织病理研究时发现硬皮病人肾损害并不少见,在通常的临床检查时不能发现。当出现氮质血症、恶性高血压、视网膜病变和高血压脑病时,患者迅速死亡,称为“硬皮病性肾危象”,是PSS的主要死亡原因。肾危象的主要受损部位在弓形动脉、小叶间动脉以及小动脉,早期以血管痉挛为主,临床上应用有力的抗高血压疗法可使肾危象逆转,血压恢复正常。晚期则主要由硬化性肾小球肾炎和肾小球毛细血管基膜增厚引起。
- 其他器官:肌肉病变除累及平滑肌、心肌外,横纹肌亦有不同程度的硬化和萎缩。患者肢体有肌肉自发痛、肌无力、肌萎缩、肌肉变硬。
周围神经和自主神经系统亦常受累,表现为末梢血循环不良,指(趾)麻木,感觉异常,掌跖发冷,多汗等。
中枢神经系统的原发性损害较少见。患者神经系统症状也可因病变累及小肠,肠壁的纤维化使肠蠕动减弱而致肠内容滞积、肠内菌群异常,因而出现营养吸收障碍,如钙吸收不良引起手足痉挛的发作,维生素812缺乏引起脊髓病变,发生亚急性联合变性。也见头发稀疏。患者可有性欲减退或消失及月经过少或闭经。
5.其他症状 除内脏病变引起的表现外,常有低热(急性发病初期可有高热)、消瘦、全身衰弱等。
实验室检查
1.常规检查:可有缺铁性贫血,外周血中嗜酸性粒细胞常增多;尿中见蛋白、红细胞、管型;血沉增快,血中纤维蛋白原含量明显增加,血液凝固性增高等。
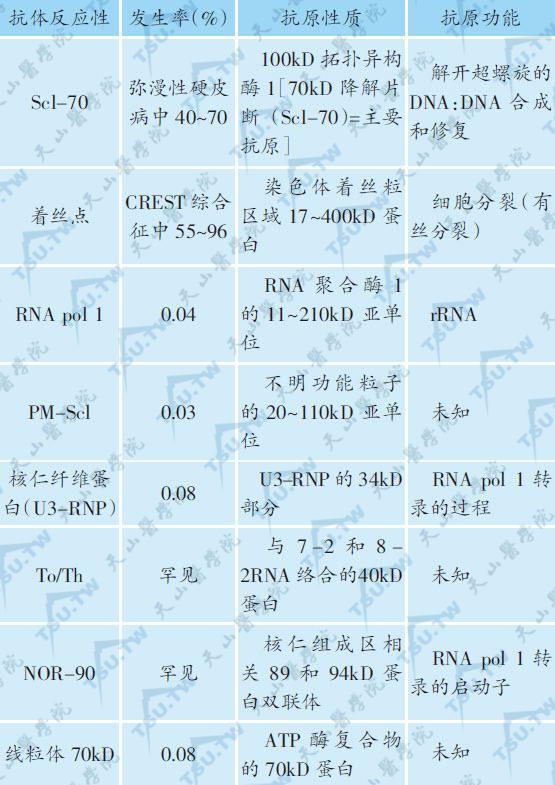
2.自身抗体:硬皮病的自身抗体见下表。间接免疫荧光测定ANA,阳性率不一,介于36%~91%之间,为斑点型和核仁型,以核仁型多见。在PSS中,抗Scl70抗体阳性率为40%~70%,特异性高,是PSS的标志抗体。在CREST综合征中,着丝点抗体阳性率为55%~96%,是该综合征的标志抗体,在弥漫性硬皮病中为8%,此有助于PSS的再分类。另外抗着丝点抗体又是硬皮病预后良好的指标。
硬皮病的自身抗体
3.其他免疫学检查 有高γ球蛋白血症和血清白蛋白降低,IgG增高,混合型冷球蛋白血症,30%~50%类风湿因子阳性,15%~70%PSS检出循环免疫复合物。
4.甲皱襞皮肤毛细血管镜检查 大多数患者显示视野模糊,有水肿,血管袢数目显著减少,血管支明显扩张和弯曲,血流迟缓,大多数病例有出血。
预后
局限性硬皮病预后较好,部分硬化性斑可自行缓解或经治疗消退,通常在消退后局部残留萎缩性瘢痕,并有色素沉着,但是线状型和致残型为进行性,通常不能消退。
系统性硬皮病的自然病程差异很大,首次确诊后的10年生存率为65%。一般为慢性进行性,但可自行缓解。病程中缓解与病情加重常交替进行。男性比女性临床过程快,且男性患者预后往往较差。有肾、心和肺受累者预后差。
本病部分患者死于肾衰竭、心力衰竭、肺部感染、营养障碍、肠坏死等。妊娠期病情可缓解呈静止状态,产后病情又再度进展。
组织病理
皮肤病理变化主要在真皮胶原纤维和小动脉。
第Ⅰ期(临床无皮损):真皮内间质水肿,胶原纤维分离及真皮上层小血管周围有轻度淋巴细胞浸润。
第Ⅱ期(临床水肿硬化):胶原纤维肿胀,血管周围细胞浸润消退,小血管及胶原纤维周围酸性黏多糖增加。
第Ⅲ期(临床硬化):胶原纤维均质化,与表皮平行排列的胶原纤维束增加,胶原纤维数量明显增多,以致向深部扩展到汗腺。弹性纤维破坏。在真皮硬化胶原内仅见少数血管,血管壁增厚,纤维化,管腔狭窄或闭塞。晚期继发改变为表皮萎缩及皮脂腺、毛囊和毛发消失,汗腺明显萎缩,周围正常存在的脂肪组织减少或消失,代之以新生胶原,还可见钙盐沉着。此外,筋膜、肌肉也可累及。局限性硬皮病改变基本上同PSS,但表皮萎缩一般不明显(图3-12-37)。
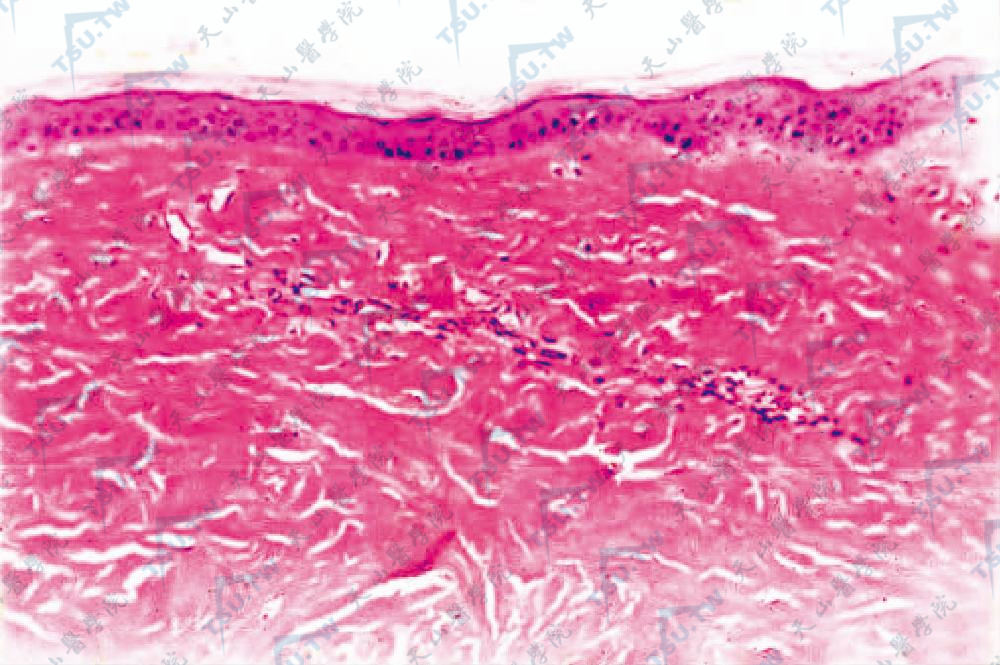
图3-12-37 表皮萎缩,真皮胶原束增粗、硬化,胶原束间有少量炎细胞(HE染色×100)
免疫病理:临床正常皮肤表皮细胞核有IgG沉积,呈斑点型或颗粒型,无特异性。少数病人皮损处的表真皮连接处有Ig沉积,但无诊断意义。
诊断及鉴别
诊断
根据局限性皮肤象牙色水肿硬化,病变活动期其周围有淡红色晕可初步诊断为局限性硬皮病。皮肤组织病理检查有助于确诊。
1.诊断标准:统性硬皮病可参考下表的诊断标准。
系统性硬皮病诊断标准(根据1980年日本厚生省特定疾病研究班制定的标准,略加修改)

2.疑诊
- 前臂伸侧皮肤活组织检查,显示本病特有的胶原纤维肿胀或纤维化。
- 除主要症状中之皮肤症状以外的其余4项症状中具有2项,并能排除其他结缔组织病。
3.确诊
- 上述疑诊病例中具有病理所见的(一)或(二)者。
- 主要症状中有3项以上者。
对于女性病人伴不规则发热,舌系带显著缩短,有弥漫性色素沉着,面、颈部及手掌呈斑纹状、多发性毛细血管扩张,并在实验室检查中有:类风湿因子阳性,抗Scl70抗体,抗着丝点抗体等自身抗体;血沉快,γ球蛋白上升,指骨末端骨质吸收或软组织钙沉着者应考虑有无硬皮病可能。
鉴别
1.局限性硬皮病应与下列疾病相鉴别
- Pasini-Pierini特发性斑状萎缩:呈不规则形、境界清楚、直径1~10cm的灰色斑,皮肤略凹陷。与硬皮病相反,本病先发生轻度萎缩,后成继发性硬化。周围无淡紫色晕。多见躯干,尤其是背部。病程多年后萎缩始终浅表性。组织学上无真皮基质硬化。但也有认为它是局限性硬皮病的一型。
- 硬化性萎缩性苔藓:其轻度硬化的斑块由有白色光泽的多角形扁平丘疹组成,斑上有毛囊性黑色角栓,有时发生水疱,最后发生萎缩。常聚集分布,但不互相融合,组织学上可与硬皮病鉴别。
- 类脂质渐进性坏死:是由红色丘疹扩展成的硬皮病样斑块,中央萎缩呈褐色且有光泽,有毛细血管扩张。病理上有特殊改变。
2.系统性硬皮病需与以下疾病鉴别
- 雷诺病:PSS早期时的雷诺现象应与雷诺病鉴别,雷诺病少有皮肤硬化或骨变化。但部分雷诺病例可能代表硬皮病的最轻型,需随访观察。
- 成人硬肿病:以皮肤深层、筋膜和肌肉的木质样变为特点。自颈部开始发病,手足很少受累,无雷诺现象及系统病变。有自愈倾向。
- 搏来霉素的皮肤毒性症状:呈硬皮病样变化,且组织变化与硬皮病相似。主要是手部的浸润斑与带状发硬,但有用药史,且为可逆性变化。
- 肢端骨质溶解症(氯化乙烯病)[aero-osteolysis(vinyl chloride disease)]:本病发生在聚氯乙烯制造业中接触氯化乙烯单体者,具有三联临床症状:雷诺现象、硬皮病样皮损和骨的溶解性损害(最常见于末节指骨),脱离接触后,部分患者皮肤硬皮病样损害可逐渐消退,溶解性骨损害亦可自然痊愈,但手指变短呈杵状。手部Ⅹ片显示手远侧指骨中心溶解性损害。
- 慢性移植物抗宿主病(CGVHD):皮肤虽有硬皮病样改变,但有接受骨髓移植病史。
- 其他:皮肌炎、混合结缔组织病、苯酮尿症、类癌综合征亦可呈硬皮病样表现,但它们有各自临床、病理、生化等方面的特点。
预防及治疗
硬皮病目前尚无特效疗法。可应用的药物及方法虽多,疗效的评价比较困难,效果也不一。
局限性硬皮病
目前多数治疗,如外用氟化糖皮质激素制剂等对局限性硬皮病都不能令人满意,一般认为本病是自限性的,多数病人几乎不需要治疗。局部皮内注射或皮损内注射中效糖皮质激素悬液的疗效尚有争议。
肢体受累病例应使用物理疗法,如音频、蜡疗、推拿、按摩等。坚持体疗、配合蜡疗能很快改善带状硬皮病的肢体关节挛缩及活动受限,恢复肢体功能。口服维生素E,每日200~300mg,有一定效果。
系统性硬皮病
1.一般治疗
- 避免精神刺激及过度紧张,注意保暖休息,避免潮湿,防止寒冷刺激、停止吸烟和避免其他诱发和加重血管收缩的因素,如应用肾上腺素、麦角新碱等,以尽量减少雷诺现象的发生。
- 除去体内慢性感染病灶。
- 尽早作维持功能的理疗及给予营养丰富的饮食等。
2.糖皮质激素 对处于病情进展期的系统性硬皮病,以及伴关节、肌肉和肺部等器官系统累及者和弥漫性硬皮病,可谨慎使用,一般常先用泼尼松30mg/d,连用数周,渐减为维持量5~10mg/d。能改善关节症状,减轻皮肤水肿和硬化及全身一般症状,对间质性肺炎和心肌病变有一定的疗效,但由于该药应用价值可疑,皮肤变软在治疗数月后才明显,而且肯定有副作用,因此不用于肢端型硬皮病,对肺纤维化和(或)有肾损害者,则应限制或不使用。
3.青霉胺 对硬皮病有一定疗效,通过干扰胶原分子间连锁的复合物,松弛分子间的结合而促使胶原纤维破坏,抑制新胶原的生物合成而发挥治疗作用,但其临床应用价值尚有争论,而且可能有严重不良反应,只限用于弥漫性硬皮病或迅速进展的肢端型硬皮病,初量250mg/d,空腹服用,每2~3个月增加一次剂量,每次增加125mg,最大剂量不要超过750~1500mg/d,18~30个月以后皮肤明显变软,维持量约300~600mg/d。最多见的不良反应为胃肠道反应,其次为肾损害,出现血尿和蛋白尿,以及白细胞和血小板减少。也有少数出现重症肌无力、肌炎、男性乳房女性化等,治疗中应注意观察。
4.秋水仙碱 可阻抑前胶原转化为胶原,或阻抑胶原的淤积,对肢端动脉痉挛和皮肤硬化有一定疗效,用量0.5~1.5mg/d,可连服数周至数月,一般疗程需2~3个月。
5.积雪苷(asiaticoside)为中药积雪草中提取的一种有效成分,实验证明能抑制成纤维细胞的活性,软化结缔组织。临床对软化硬皮、消除组织水肿、缓解关节疼痛、愈合溃疡等均有相当效果。片剂(每片含积雪苷6~10mg),每次3~4片,1日3次;积雪苷针剂(每支2mL,含积雪苷20mg)肌内注射,每周2~3次,每次1支,一般1个月左右开始见效。
6.血管痉挛的治疗
- 血管扩张剂:外用血管扩张药如1.2%烟酸苄酯霜,1%~2%三硝酸甘油软膏。口服血管扩张药如肼屈嗪25mg每日3次,也可用地巴唑、妥拉唑林。加入8~16mL丹参注射液(每毫升相当于原生药2g)的低分子右旋糖酐静脉点滴,每日1次,一疗程1个月,连续或间歇使用,对系统性硬皮病早期有较好疗效,且血沉和IgG均可转为正常。亦可用丹参注射液肌内注射,每次4mL,每日1~2次,有一定的疗效。此外也可口服丹参片。
- 增强纤维蛋白溶解:如司坦唑醇(stanozolol)2mg,一日2~3次,一疗程6个月左右。
- 抗血小板凝固药物:如阿司匹林300mg,一日2次。
- 其他抗血管痉挛的药物:前列腺环素、硝苯地平(nifedipine)、盐酸哌唑嗪(prazosin)、雌三醇(estriol)等。亦有人用利血平1mg加入5mL生理盐水缓慢注入肱动脉,可减轻疼痛。
7.局部治疗 发生指部溃疡时需局部清创,切除纤维和脓性物,油纱布包扎加之抗生素和止痛剂。疼痛性钙化结节可外科切除。
8.血浆置换 严重PSS,有报道试用血浆置换疗法。
9.中药 主要用活血化淤药,可改善微循环及结缔组织代谢。
对于久病体虚,合并症较多,临床症状复杂或经长期治疗而效果缓慢不易巩固者,则除选用合适的活血药外,必须强调辨证施治,对体虚患者尤需注意调治。
对病情顽固者剂量一般宜大,服药时间要长,可选用下方:
- 解毒活血汤(首乌15g,鸡血藤25g,元胡12g,乳香、没药各6g,泽兰25g,丹参21g,夏枯草15g,元参21g,郁金12g,血竭6g),气血亏加黄芪、桂枝、当归、白芍等;阳虚者加附子,肉桂。
- 脾肾阳虚者用阳和汤加减。
- 仙茅、仙灵脾各9g,生熟地各9g,桂枝、红花各9g,全当归15g,赤芍9g,炙草3g。
- 右归丸、成药附归八味丸或全鹿丸均6g,任选一种,日服3次,温开水送服。
- 软皮丸(川芎、炮姜、桂枝、丹参、桃仁、当归各30g,共研细末,炼蜜为丸)每次9g,每日2次。
- 黄芪、党参、当归、丹参各15g,赤芍、川芎各9g,红花、桂枝各6g,鸡血藤9g,肉桂3g,仙灵脾、蝮蛇、祁蛇各9g,甘草6g。
- 鸡血藤片口服。
- 虎骨酒或红灵酒外搽。
