
汗孔角化症(Porokeratosis of Mibelli)是一种较少见的、起源于遗传的慢性进行性角化不全性皮肤病。以边缘堤状疣状隆起、中央轻度萎缩、组织学上存在角质样板层(cornoid lamella)为特点,由Mibelli于1893年首先报道并命名。Mibelli认为本病的损害是由于汗孔部角化障碍所致,因此命名为汗孔角化症。但有些学者发现皮损边缘的角质样板层可能和毛囊有关,并且病损也可见于黏膜、龟头及其他无小汗管的部位,故这一名称可能是错误的。
Mibelli于1893年描述了经典型汗孔角化症的皮损,同期Respighi和Andrews独自描述了其异型浅表播散型。线状型在20世纪的早期被报道。1966年Chernosky描述了播散性浅表性光线性汗孔角化症。1977年又报道了点状汗孔角化症,它通常是线状或Mibelli型的变型。
病因及发病机制
汗孔角化症属常染色体显性遗传,可在一个家族几代成员中发病,如林达等(2003年)报道一家系4代26人中有10人患病,每代均有患者,且无性别差异,患者必有双亲之一患病,患者的同胞中均有38%患病,连代遗传。但也有无遗传证据而散发于人群者。角质形成细胞发育不良(keratinocyte dysplasia),Otsuka等报道在皮损区角质形成细胞呈非整数倍和常染色体异常,肿瘤抑制蛋白P53有过度表达。
也有报道成纤维细胞,特别是常染色体3有细胞遗传学异常。已证明该病是表皮的细胞不正常的克隆增生,这些增生可受某些刺激而激发,尤其是免疫抑制剂,其他如肾移植、电子束辐射、光化学疗法、日光及慢性皮损损伤等均可诱发或加剧皮损,故认为免疫抑制在有遗传倾向者中可直接激发表皮突变克隆的表达或破坏表皮生长动力学而促发异常克隆的增生,从而诱发本病。此外,Inamoto等报道某些药物如苯甲基双氯噻嗪也可诱发本病。对于器官移植、接受免疫抑制治疗的病人、HCV及HIV感染者中本病的高发生率,有人用感染因素来解释。
临床症状
皮损开始为一小的角化性丘疹,缓慢地向周围扩展形成环形、地图形、匐形性或不规则形的边界清楚的斑片,边缘呈堤状、有沟槽的角质性隆起,灰色或棕色,中心部分皮肤干燥光滑而有轻度萎缩,缺乏毳毛,其间汗孔处有时有针头大细小的角质栓。皮损形态不一,可从细小的角化性丘疹直至巨大疣状隆起,有时因边缘窄,颜色深而像一圈黑线,或因向单一方向扩展形成线状,或因中央发生新疹而形成多环形。皮损直径大小可自几毫米至几厘米,数目也因人而异,从单个至百余个不等,数目多时常呈带状分布于某一区域,受外伤处可以出现新疹。
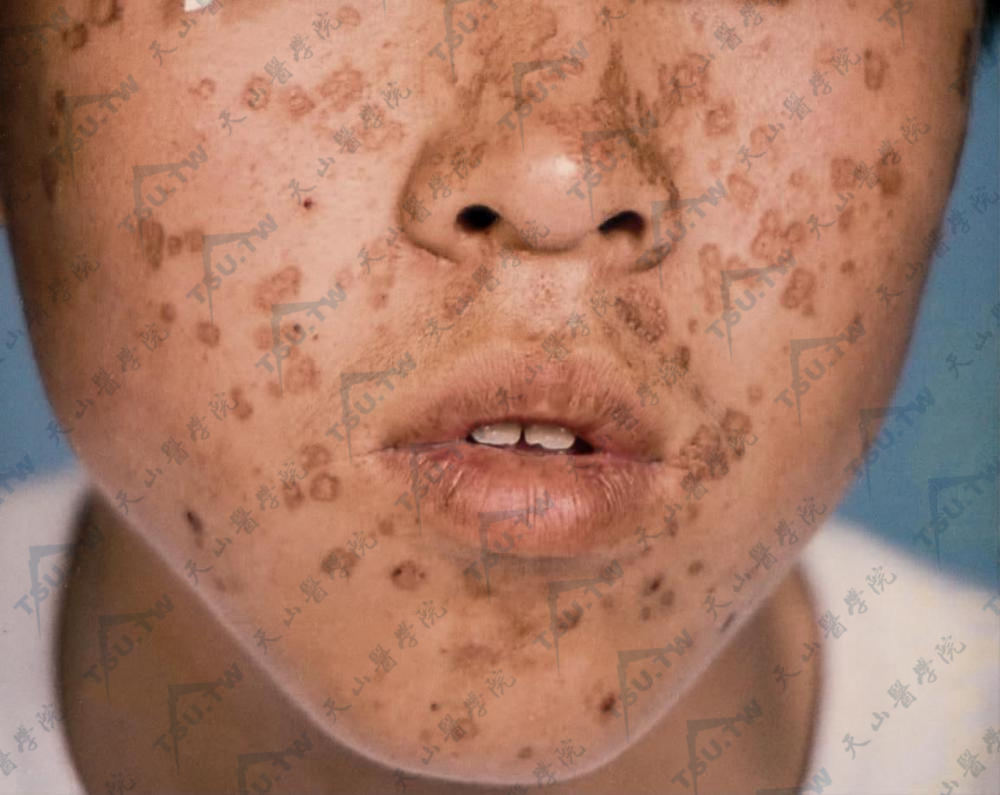
图:面部见多数环形、不规则形边界清楚的斑片,边缘窄,呈线形堤状角质性隆起,灰色或棕色,中心部分有轻度萎缩
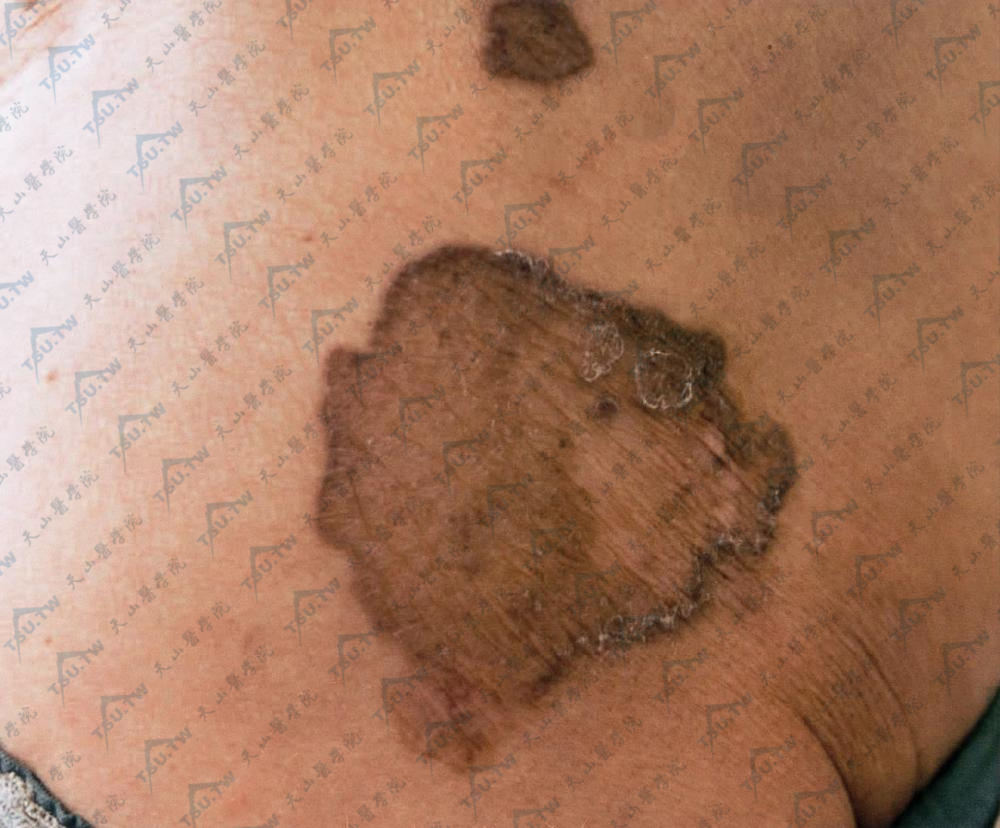
图:躯干部典型损害,呈圆形或不规则形斑片,边缘堤状隆起,黑褐色,中心部有W陷、萎缩
皮损好发于四肢(尤其在手、足部)、面部、颈部、肩部及外阴,也可累及头皮及口腔黏膜,不同部位的皮损有不同的临床表现,位于受压或摩擦部位皮肤增厚处者,堤状角质性隆起的边缘特别显著;位于趾间者类似鸡眼;位于面部者边缘为一圈黑线而隆起不显;位于皮肤娇嫩处(如腋下),其角化和萎缩均轻;位于踝部者皮损有时类似疣状痣;位于头皮者产生斑秃;位于口腔黏膜者边缘浸渍,呈乳白色升高的条索;位于阴茎者产生糜烂性龟头包皮炎。如甲母质受累则可发生甲营养不良、甲板增厚、浑浊并起嵴纹。
本病男性较多见,初发于幼年期,但也有起于成年期者,一般无主观症状。皮损往往持续存在,趋向缓慢和不规则的进展。
汗孔角化损害易于恶变,恶变大多发生在线状型,且大多数在下肢。Goerttler报道巨大汗孔角化症恶变率为7.5%~11%。Jame等报道了32例癌变病人的情况,其中21例发生鳞癌,8例为Bowen病,3例为基底细胞癌。
病程和预后:汗孔角化症随时间皮损增大和数目增多。虽然这在Mibelli汗孔角化症中进展非常缓慢,但在DSP中进展是很显著的。DSAP病人在紫外线照射下的皮疹和由药物所诱导的皮疹类似。在免疫受损的病例中,病情的波动可能与免疫状态相平行。所以DSP或DSAP突然恶化应该立即寻找潜在的免疫抑制。恶变(鳞癌,Bowen病,基底细胞上皮瘤)常发生于Mibelli和线状汗孔角化症大的、孤立的长期皮损,但它也发生于DSP、DSAP和PPPD。据统计,日本病人发病较早,在汗孔角化症皮损中更易发生多发的恶性肿瘤。
本病除了如Mibelli所述的经典斑块型汗孔角化症(常为单个或仅数个孤立性角化损害,主要分布在手足、前臂和大腿等处)外,还有一些异型,主要有:
- 浅表播散型(DSP):多见于面、颈、前臂、躯干及掌跖,边缘纤细如一圈黑线,中央有色素沉着,类似萎缩性扁平苔藓。
- 单侧线状型:皮损类似疣状线状表皮痣,常单侧分布。本型可能是线状苔藓的一个型或为非遗传性,起病于婴儿期并可自愈的特殊类型,或为外伤所致的同形反应现象。
- 播散性浅表性光线性汗孔角化症(DSAP):见后述。
- 显著角化过度型:皮损发红,中心区增厚及边缘角化过度明显,其他同经典斑块型。
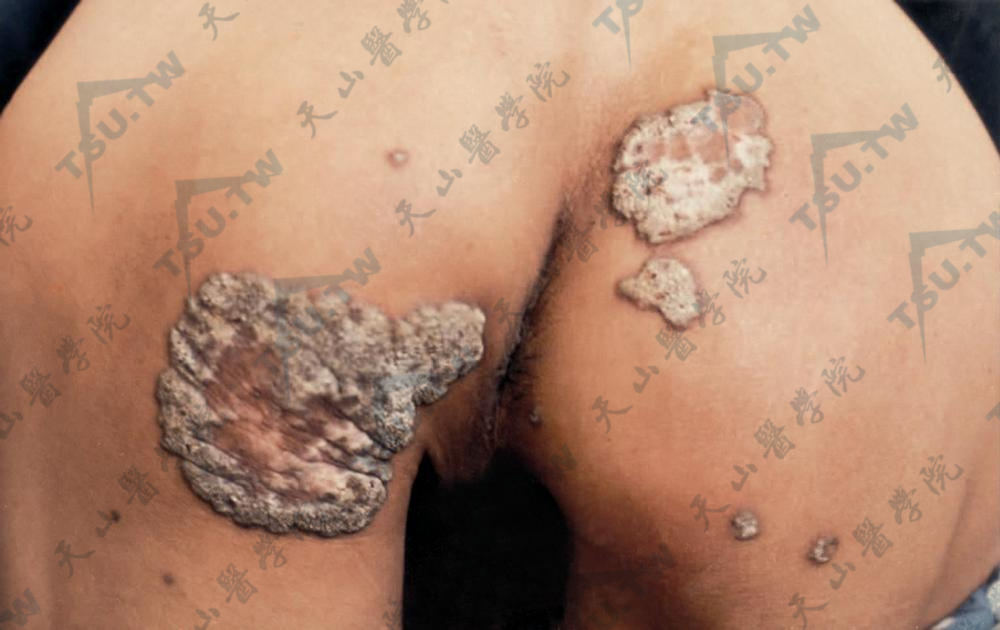
皮损中央及边缘增厚,角化过度,呈疣状隆起,仍可见堤状隆起边缘
- 炎症角化型:皮损类似老年角化症,并可发生溃破、结痂的增生性炎症反应而使外观似鳞癌。
- 掌跖泛发性型(PPPD):好发于男性,与日光无关,首先在掌跖部发生皮疹,以后泛发到全身。
- 点状汗孔角化:症多在儿童或青春期发病,为1~2mm大小点状角化性或棘状丘疹,少数皮疹为3~5mm的角化性丘疹(见后述)。
- 丘疹型:为2~4mm大褐红色形态不规则的丘疹,可有融合现象,部分区域可见汗孔角化症的典型损害。
- 疣状斑块型:多见于青壮年臀部等易受压或摩擦部位,表现为疣状增生性丘疹或斑块,皮损面积大,遗传史可不明确,但有特征性组织病理——角质层内可见由角化不全细胞组长的“鸡眼样板”,其下方的颗粒层减少或消失,棘层内有角化不良细胞。
- 混合型:同一病人除有经典斑块和浅表播散型皮损外,还存在结节等缺乏本病特征的皮损。
组织病理
取自周围高起的角化过度嵴处,镜下该嵴呈充有角蛋白的凹陷。其凹窝以某种角度向下方深部伸展,其顶端偏离损害中央部位。在充有角蛋白的凹窝部中央,有一不全角化柱,即圆锥形板层,此为本病最有特征的组织相。在板层内,角质细胞呈同质性并有嗜碱性固缩核,其下方的表皮,角质形成细胞排列不规则,胞核固缩伴核周围水肿。皮损中央部位的表皮棘层萎缩,但也可呈正常厚度(偶有肥厚),真皮可见非特异性血管周围慢性炎细胞浸润。
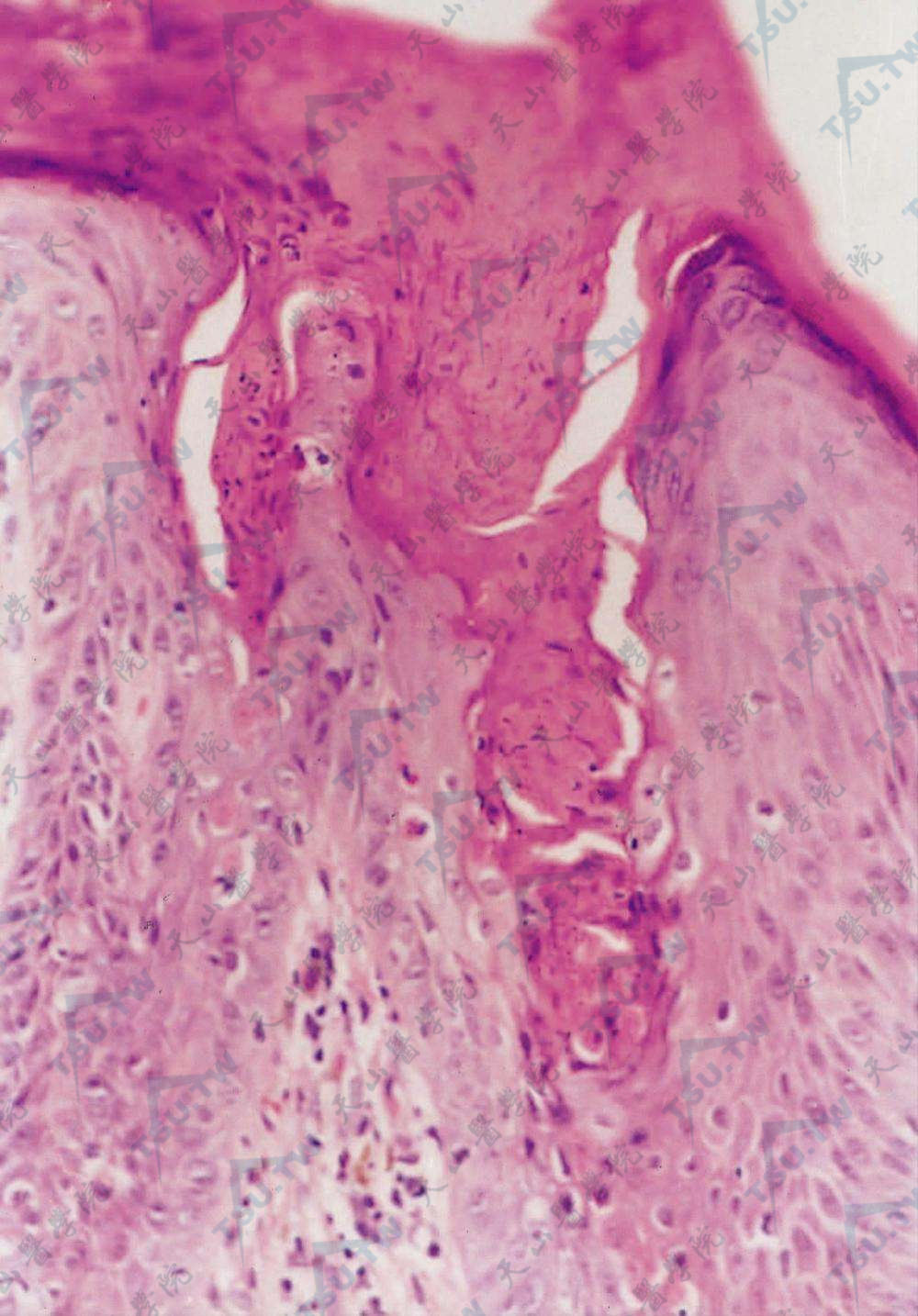
见角化不全柱,呈“鸡眼样层板”(HE染色×400)
电子显微镜检查可见角化不全柱下方表皮的许多角质形成细胞变性,表现为核固缩,胞质有大的空泡,而其周边可见张力丝凝聚,在角化不全柱基底部可见残核和张力丝簇构成的角化不良细胞,角化不良柱主要由固缩核以及由于出现许多部分退化的细胞器而具有高电子致密度的胞质所构成。
诊断及鉴别
本病根据临床表现,一般诊断不难,必要时可作活检以证实,因本病的组织病理象有诊断价值。
在鉴别诊断方面,本病需与扁平苔藓、萎缩性硬化性苔藓、疣、光线性角化症、疣状表皮痣、Bowen病、环状穿通性肉芽肿、环状晚期梅毒疹、环状弹性纤维溶解性肉芽肿以及匐行性穿通性弹性纤维病相鉴别。
预防及治疗
各种治疗方法疗效均不满意,任何治疗方法都不能防止其复发。
可外用10%水杨酸软膏或0.05%~0.1%维A酸软膏,外用氟尿嘧啶封包治疗对本病是一种简单而有效的方法。如McDonald等采用5%氟尿嘧啶霜或溶液在皮损部作封包,直至皮损产生明显炎症甚至溃疡,结果发现约经3周即可获痊愈。对过度角化的角质使用角质溶解剂治疗通常能减轻症状。
内服阿维A酯、阿维A或异维A酸往往在用药期有效,如Danno报道1例播散性汗孔角化症病人用阿维A酯每日1mg/kg,治疗2周后开始见效,3个月后疗效更显著,但在停药后趋向复发。对疑与日晒有关的病人,可试服氯喹治疗。
对局限性皮损可用CO2激光、电灼、液氮冷冻或作手术切除。
鉴于免疫抑制剂可促进汗管角化症皮损的发展,近期国外有人用5%咪喹莫特乳膏(有促进Th1型细胞介导的免疫反应作用)局部封包治疗1例Mibelli型病人取得了很好的疗效。
因本病的损害位于表皮和真皮浅层,故也可试用皮肤磨削术来治疗,但磨削的深度不能超过真皮乳头层,否则术后会遗留瘢痕。
由于本病可有恶变,故对于局限性的皮损均应予以切除或破坏;对播散型病人应接受定期随访,遇有在角化斑基础上发生增生损害时应及时做活检,一旦有癌变趋势即应切除、冷冻或作电灼、激光去除,以防后患。
