
本病又名假性毛囊角化不良病(dyskeratosis pseudofolliclaris),系1889年首先由Darier命名,故又称达里埃病(Darier's disease)。本病是一种少见的,以表皮细胞角化不良为基本病理变化的慢性角化性皮肤病。因本病损有融合、增殖的倾向,故又曾被称为增殖性毛囊角化病(keratosis folliculalaris vegetans)和增殖性毛囊角化不良病(dyskeratosis follicularis vegetans)。
病因及发病机制
本病是一种常染色体显性遗传引起的角化过程异常的遗传性皮肤病。日高统计一组222例患者中99例有家族史。患者有桥粒斑蛋白(desmoplakin)的异常,特别是桥粒斑蛋白Ⅰ和Ⅱ,桥粒珠蛋白和桥粒芯糖蛋白的异常溶解。水疱形成是由于在张力细丝与桥粒附着处有缺陷的结果。在本病患者的棘层松解细胞中钙依赖性细胞黏附分子(上皮钙黏素)显著减少。基因定位于12q23~24.1,并编码钙依赖性ATP酶(SERCA2)。
它是由于染色体12q24.1的ATP2A2基因突变引起,该基因编码肌浆网和内质网的钙离子2型ATP酶(SERCA2),SERCA2是使得内质网中大量钙离子聚集的离子泵之一。它有两种异构重整型(isoforms):SERCA2a表达于心肌和平滑肌中,而SERCA2b表达更广泛,还包括表皮中。本病是由两者无意义表达和插入和(或)清除突变引起,有可能导致单纯功能不全和错义突变,产生非正常表达的蛋白。
除遗传因素外,有些学者根据部分病人血清中维生素A浓度低及应用维生素A治疗有一定的效果,因而认为本病与维生素A代谢障碍有关,但意见不一,故有关维生素A在本病病因学中的作用仍需进一步研究。由于本病病理上有角化不良的特征,故可认为本病为表皮合成和转换障碍的家族性疾病。Caulfield等电子显微镜的研究已显示有张力丝-桥粒复合物缺陷,这些缺陷可以是此复合物合成、结构或成熟上的缺陷。Rubin报道有因服用碳酸锂而诱发本病者。
在1981年,Jegasothy等报道了本病的另一体征,在他们观察的8例病人中,7例病人有无反应性(anergy)和不能产生白细胞抑制因子,病人的淋巴细胞对刀豆球蛋白A完全无反应(但对植物血凝素和美洲商陆分裂素反应正常),这一发现促使对本病的免疫系统改变作更多的研究。目前认为,因为病人有无反应性及本病有角质形成细胞的损伤,故易合并由单纯疱疹病毒所致的Kaposi水痘样疹。
由于病变在夏季出现,曝光区域严重及日晒后加重,故日光被认为是重要的致病因素,但病损也见于口腔黏膜、腹股沟、掌跖部等非曝光部位,而这些部位常易受直接的物理创伤,故非日光性损伤也可在皮损的发生中起重要作用。
临床症状
本病出生时没有,通常开始于10~20岁。男女无差异。好发典型部位为面部前额头皮和胸背。这些部位有很多皮脂腺,但皮损也发生于无皮脂腺部位(掌跖)、角化和无角化上皮如黏膜、角膜和下颌下腺。本病见于各种族,男女发病率相等。患病率为1/50000~1/100000,通常儿童期开始发病。
早期的皮损为细小、坚实、正常肤色的小丘疹,但不久即有油腻性、灰棕色、黑色的痂覆盖在丘疹顶端凹面,去除后丘疹顶端暴露出漏斗状小凹,丘疹逐渐增大成疣状,常群集并趋向融合,形成不规则的疣状斑块。位于屈侧腋下、臀沟及阴股部等多汗、摩擦处的损害增殖尤为显著,形成有恶臭的乳头瘤样和增殖性损害,其上有皲裂、浸渍及脓性渗出物覆盖。
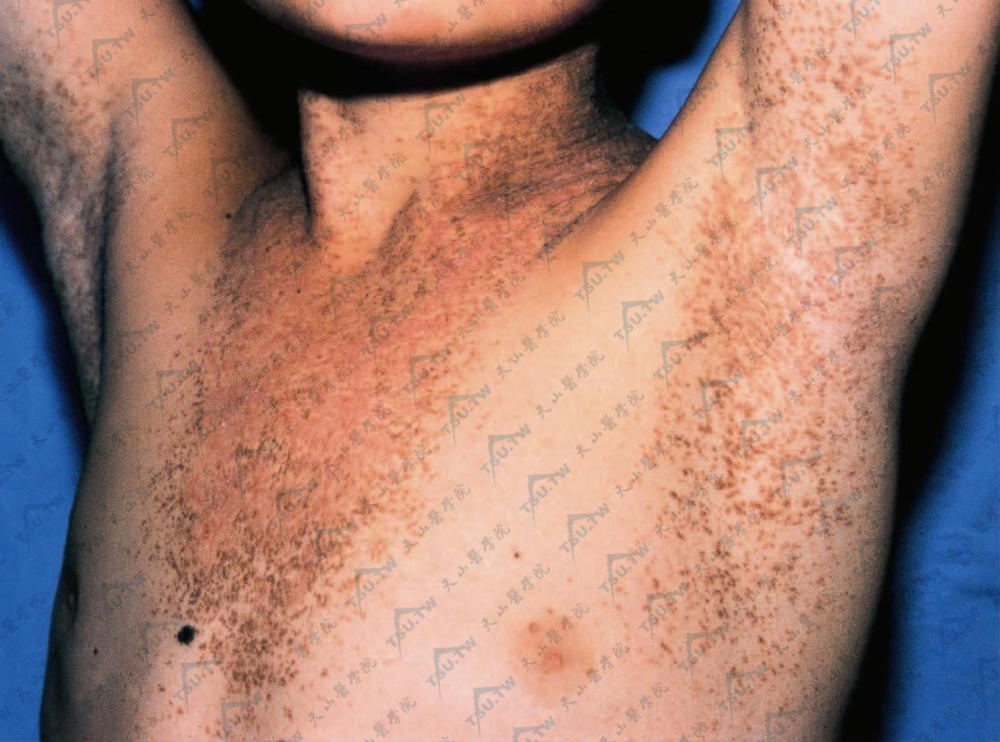
颈、颈前三角及两腋见多数毛囊性小丘疹,增大呈疣状,色棕黄、污灰,顶端覆以油腻状黏着性痂或糠样鳞屑
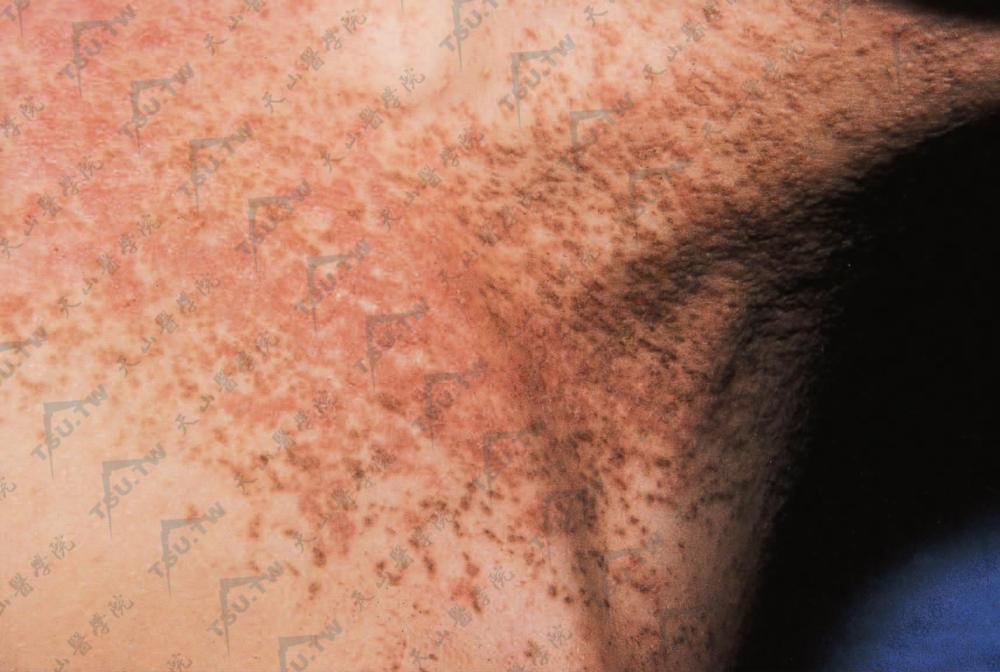
与上图为同一患者的腋部见毛囊性疣状丘疹,色棕黄、污灰,部分可融合成不规则斑块
皮损好发于皮脂溢出的部位,如头皮、前额、耳、鼻唇沟、颈、肩、前胸、背中线部、腋下等,也可扩展到整个躯干、四肢屈侧、臀部和生殖器部,最早皮损的常见部位是耳后。头皮部的皮损常覆盖油脂样污痂,一般无脱发。面部的皮损在鼻部特别严重,唇部可有结痂、皲裂、肿胀和浅表性溃疡,舌背部可发生斑状角化和浅表性糜烂,在齿龈和腭部常可有小白丘疹;在掌跖常可有点状角化,并可相互融合形成掌跖弥漫性角化;在手足背和胫前可有扁平疣样丘疹,往往呈线状排列,或可类似为疣状肢端角化症(Hopf),消退后可遗留白色斑点;指甲可发生甲下角化过度,甲脆弱、碎裂、白色或红色纵纹,甲游离缘有三角形缺损。Ronchese认为本病的指甲变化具有特征性,对早期或症状不典型患者的诊断有帮助。偶尔在手足上可见到出血性丘疹,这似乎代表着一种基因变异,因为它们是家族性发生,可以由特异突变引起。非典型的或异常严重者通常是错义突变的结果。
本病还可累及口咽、食管、喉和肛门直肠黏膜。
皮疹通常是对称和广布的,但明显的不对称或带状、痣样分布也可以发生,有些病例皮损部可发生大疱。在局限性毛囊角化病,皮疹沿着Blaschko线局限性或带状分布,躯干为其好发部位,大多呈线状,但当发生于别处时诊断困难,需依通过皮损活检来确诊。
本病常在夏季加重,患者对热敏感。严重日照后可诱发本病(photo-Köebner反应)。UVB亚红斑量照射可在某些病例中复制出皮损。大多数病例冬季好转或痊愈。患者因皮肤糜烂而常感严重瘙痒和不适,也可致疼痛,易渗血和产生强烈臭味。碳酸锂在某些人中可诱发本病。
本病实际上并不是一种毛囊性疾病,这可由某些缺乏毛囊的部位,如掌跖和口腔黏膜可以发病的事实得到证实,故毛囊角化病这一名称易造成误解。
组织病理
本病的特征性病理改变为:
- 特殊形态的角化不良,形成圆体和谷粒;
- 基底层上棘层松解,致形成基底层上裂隙和隐窝;
- 被覆有单层基底细胞的乳头,即“绒毛”向上不规则增生,进入隐窝和裂隙内;
- 可有乳头瘤样增生、棘层肥厚和角化过度,真皮呈慢性炎症性浸润。
圆体见于Malpighi上部层,特别是颗粒层内,其中央为均质性嗜碱性固缩核,核周绕以透明晕,在晕周围有像壳一样的嗜碱性物包绕;谷粒则见于角层和隐窝,类似角化不全细胞,但个体稍大,常呈谷粒状,周围继以均质性角化不良物质,后者染色常呈嗜碱性;隐窝为小的裂隙样表皮内水疱,常直接位于基底层上方,其中棘刺松解细胞呈部分过早角化。
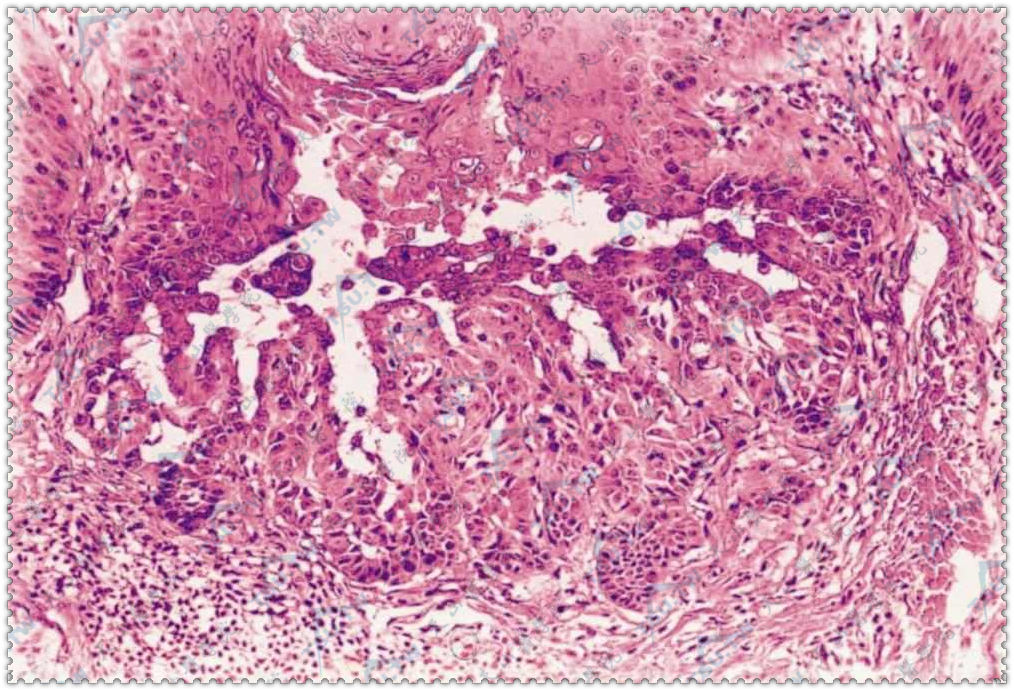
表皮基底层上裂隙,裂隙底部绒毛生长(HE染色×100)
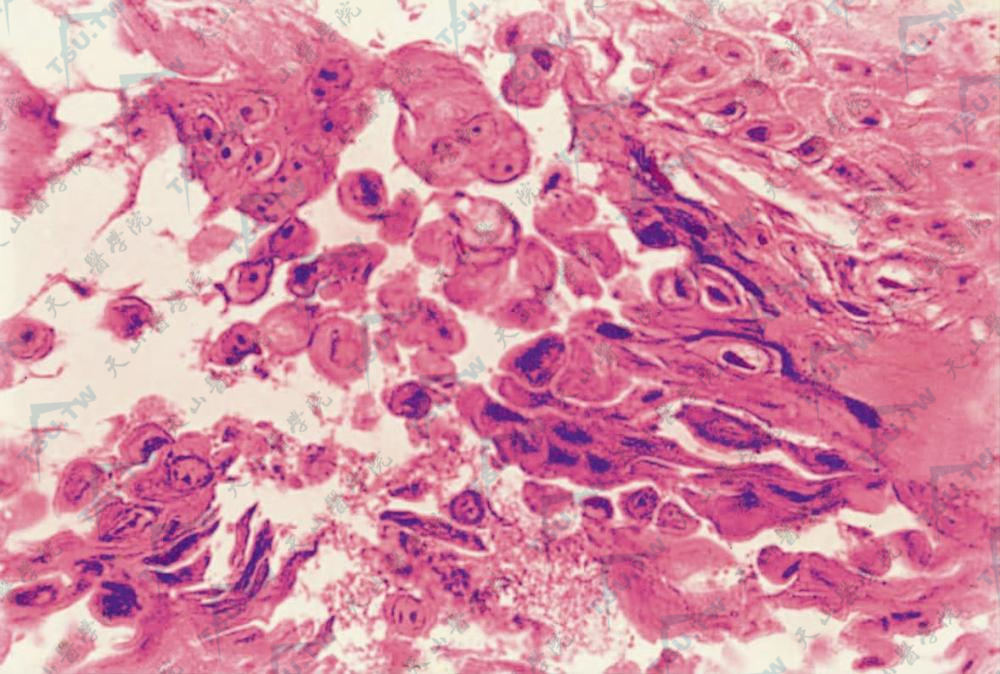
左下角为表皮角化不良细胞(圆体和谷粒细胞)(HE染色×400)
口腔黏膜损害的组织相与皮损相似,也有角化不良和隐窝,但一般无典型的圆体形成。
诊断及鉴别
根据临床表现及病理检查结果,诊断不难,但有时需与以下疾病相鉴别:
- 黑棘皮病 皮损色深,多局限于腋下、腹股沟等身体屈侧部位,呈柔软的乳头瘤状,恶性型常合并内脏腺癌。
- 融合性网状乳头瘤病 青年期发病,好发于两乳房间、双肩胛间,为黄棕色扁平丘疹,并逐渐融合成网状斑片。
- 脂溢性角化 好发于中年以上成人的面部、手背、躯干和上肢,为褐色扁平斑丘疹,表面光滑或呈乳头瘤样改变。
- 暂时性棘层松解性皮病 中年人多见,好发于躯干,组织病理上可有毛囊角化病的表现,但皮损以丘疹、丘疱疹为特征,皮损一般可在数月内自行消退。
- 疣状角化不良瘤 常为头部或颈部的单个疣状结节。
- 在组织病理上应与下列疾病相鉴别:①日光性角化病,常有表皮细胞核的间变;②慢性良性家族性天疱疮,表皮无裂隙,而有基底层上的棘层松解的大疱。
预防及治疗
轻症病人无需治疗,可以局部使用润滑剂,注意卫生。维生素A已被应用多年以减轻病情,虽有获得不同程度成功的报道,但多数效果并不理想,常规用量为5~10万U/d,但用30~100万U/d成功率更高些,在用大剂量时往往有维生素A过多的副作用;Ayres等用维生素A 15万U/d加维生素E联合应用能获效。氯喹0.25g,每日1~2次,或羟氯喹0.2g,每日1~2次,也有效。
近年来已逐渐用维A酸来治疗本病并替代维生素A。对于局限型,外用维A酸即可生效。他扎罗汀和阿达帕林亦可能有效。系统应用维A酸可用于严重病例。其作用机制是促进DNA合成,加速生发层的有丝分裂速度,使上皮细胞正常化、颗粒层增厚、角化不全减少,使角化不良恢复正常。比较有效和常用的合成维A酸有三种:异维A酸(isotretinoin)、阿维A酯(etretinate)及其主要代谢产物阿维A(acitretin)。Orfanos用阿维A酯50~75mg/d治疗能迅速控制皮损症状,以后逐渐减量改用维持量25~30mg/d。应用异维A酸每日0.5~1mg/kg、阿维A 25~30mg/d或我国研制的维胺酯25mg,每日3次也能获效。一部分病人会发生严重的炎症,用环孢素治疗有效、可控制严重发作。继发性金黄色葡萄球菌感染与皮损加重有关,应积极予以治疗。
对炎症皮损的局部治疗包括外用糖皮质激素霜或软膏,也可外用水杨酸、煤焦油或硫磺软膏等。Fulton等用0.1%维A酸霜封包治疗数月可使皮损痊愈;局部应用氟尿嘧啶治疗对本病也有一定帮助。对小斑片损害,用曲安奈德局部注射可使皮损获得迅速但暂时性的缓解;对肥厚性皮损,擦皮术、冷冻、激光或切除后植皮等均可予考虑。浅层Ⅹ线或境界线照射可改善症状。
病人应避免烈日曝晒。保持局部清洁,减少局部摩擦。由于本病为遗传性疾病,故应绝对禁止近亲结婚。
