
本病是1901年丹麦Ehlers和1908年Danlos首先系统完整描述的,故也称为埃勒斯-当洛斯综合征(Ehlers-Danlos syndrome,EDS)。其他命名还有弹性皮肤(cutis elastica)、皮肤毛细管破裂(dermatorrhexis)等。
本病为一种具有遗传倾向的疾病,主要影响结缔组织。临床上易受累的组织和器官包括皮肤、关节、心血管系统、胃肠道和眼。病情程度不等,从轻度到致死性病变。遗传方式多样,可为常染色体隐性、常染色体显性或性联隐性遗传。本病相对少见,迄今报道的病例不超过200例。
病因及发病机制
在狗和猴身上形成的实验模型和人类的Ehlers-Danlos综合征很相似,多以常染色体显性方式进行遗传。特征性的临床表现为皮肤脆性增加、皮肤松弛和皮肤过度伸长。组织上真皮中胶原纤维和弹性纤维的大小、形状和排列方向均有改变。部分纤维破碎,基质和酸溶性胶原纤维也有增加,测定患病动物皮肤,其拉长度可减少到正常狗的1/27或正常猴的1/3。
尿中羟脯氨酸和黏多糖的含量在正常范围之内,用层析法检测尿中的氨基酸未发现代谢异常,也未发现染色体和血液学方面的稳定性异常。有人认为皮肤的伸长度有某些异常改变与胶原纤维排列异常有关。Varadi和Hall未发现患者皮肤中的氨基酸成分、锁链交互连接和弹蛋白的量有何改变,他们认为由于过度松弛发生在关节囊和筋膜,而这些部位的组织几乎全部由胶原纤维构成,因此认为本病胶原纤维的缺陷比弹性纤维更为突出。
结缔组织的胶原蛋白可分为六型,它们有特定的分布部位,在基因和生化方面既密切相关,又各有特点。正常人体皮肤中主要为Ⅰ型和Ⅲ型胶原,Ⅰ型占成人胶原总重的80%,Ⅲ型约占15%,其余5%为Ⅳ型、AB型和Ⅰ型的三聚物,Ⅱ型主要分布在软骨中,各型的确切功能尚不清楚。
最近研究证明,可用Ⅰ型胶原和Ⅲ型胶原代谢紊乱作为基础来对本病的发病机制进行解释。根据Beighton等报道,本病患者的Ⅰ型胶原、型胶原和Ⅲ型胶原的生化缺陷非常明显,Ⅲ型胶原的合成减少或合成过程不正常;患者的赖氨酸羟基酶存在缺陷,导致羟基赖氨酸衍生物交互连接减少。Lichtenstein等发现患者Ⅰ型胶原中的NH2终末伸展部分裂开,这种缺陷类似牛和羊的以隐性遗传方式的Ehlers-Danlos综合征。此外,患者的血浆中纤维连接蛋白(fibronectin)也存在着功能上的缺陷,从而改变了血小板的凝聚性能,但它不导致结缔组织的变化。
临床症状
本病的特点是皮肤和血管的脆性增加,皮肤弹性过度,关节伸展过度。此外,还可能合并骨、眼、内脏病变以及其他先天性畸形。症状最早发生于幼儿,常常在开始学走路时表现出症状,随后症状逐渐明显。轻型患者症状常被忽略。两性均可累及,男性发病率较高。

指关节伸展过度
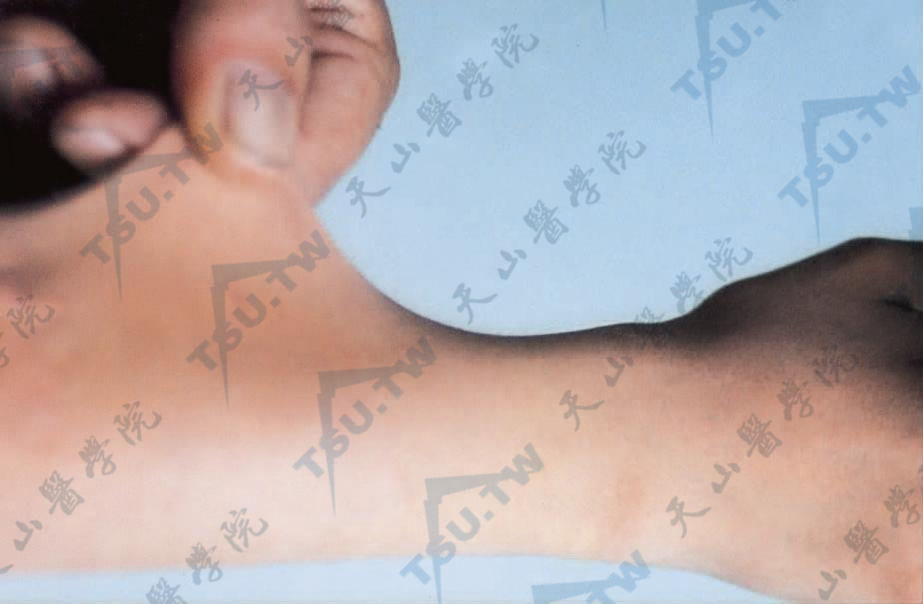
拉起皮肤时可过度伸展,放松后可恢复原状。皮肤柔软,摸之有绒样感
外伤所引起皮肤损伤在程度上不相称,一个轻的外伤可造成一个大的伤口。愈合过程也较慢,并留下大的萎缩性瘢痕。有时在此瘢痕上形成蓝灰色的海绵样瘤。皮肤于轻度碰伤之后,可引起明显的血肿。伤口缝合后容易反复裂口。
皮肤弹性过度,其程度和范围不同。在褶皱处拉起皮肤然后放松时,皮肤可恢复其原来位置。皮肤柔软,摸之有绒样感。妊娠之后一般不引起萎缩纹。
关节伸展过度,其程度也不一样。轻者仅限于指间关节,重者患儿步态困难,易摔跤。四肢大关节在轻度外伤作用下即可引起半脱臼,有时发生自发性半脱臼。可伴膝关节反屈和脊柱后侧凸。
患者的面部也有一些特征表现,如眼距增宽,鼻背宽而平,眼内眦形成皮赘。老年患者眼四周皮肤丰满而多皱纹。此外,小腿胫部和上肢的前臂可见许多豌豆大小的皮下结节,质地较硬,皮肤不红肿。有时伴有肢端青紫现象。
其他症状包括:眼部常有血肿形成,巩膜呈蓝色,眼底有血管纹。由于胶原纤维减少和肌张力差,可引起单发性或多发性疝,如脐疝、膈疝、腹股沟疝等。可引起肠胃道的单发性或多发性憩室和膀胱憩室。主动脉、颅内动脉及其他部位的大血管可发生单发性或多发性动脉瘤,动脉瘤破裂后可引起急性大出血或其他致命性并发症。还可伴有先天性心血管畸形、先天性肾脏发育不全等。
部分患者可伴有其他先天性疾病,如成骨不全(osteogenesis imperfecta)、弹性纤维性假黄瘤和Marfan综合征。
有人根据本病的临床表现、遗传方式以及生化改变,将本病分为:
10种亚型
Ⅰ型(重型):常染色体显性遗传。临床表现为关节活动过度,皮肤伸展过度,皮肤严重撕裂,假性肿瘤,骨骼肌所致的畸形,静脉曲张,皮肤易碰伤等。此外,羊膜早破及早产儿的发生率也很高。
Ⅱ型(轻型):常染色体显性遗传。临床表现为轻度关节活动过度,皮肤对碰伤和撕裂的耐受较好,假性肿瘤的发生机会少,早产的发生率不高。
Ⅲ型(良性关节活动过度型):常染色体显性遗传。本型又包括了许多所谓“不全型”。临床上表现为全身关节活动过度,皮肤不同程度伸展过度,有轻度皮肤易撕裂倾向,其他并发症少见。
Ⅳ型(淤斑或动脉型):常染色体显性或隐性遗传,为Ⅲ型胶原蛋白合成减少或不正常。临床表现为关节活动过度,但仅限于指间关节,皮肤伸展一般不明显或很轻,肌肉骨骼引起的畸形不是本型所特有。另一特点是一个很轻的外伤可引起明显的血肿。此型病人易患严重心血管和胃肠道疾病。可因动脉破裂和大出血导致死亡。
Ⅴ型(性联型):首先由Beighton(1969年)报道,患者轻度关节活动过度,但皮肤伸展过度很显著,中等伸展则易使皮肤撕裂和碰伤。肌肉骨骼并发症和假性肿瘤常见。
Ⅵ型(眼型):常染色体隐性遗传。由于赖氨酸羟基酶缺陷,导致羟基赖氨酸交互连接减少。临床特点为Marfanoid素质,皮肤软、薄、丝绒状,易形成瘢痕。细长指,脊柱向后向侧弯畸形,中等关节活动过度。眼病变为本病特征,表现为眼易受损伤,并有锥形角膜、青光眼和小角膜。
Ⅶ型(先天性多发性关节松弛型):可能为常染色体显性遗传。由于Ⅰ型胶原蛋白的NH2终末延伸部分裂开。临床特点是患者具有特征性面孔,身材矮小。由于明显的关节活动过度而引起多发性关节脱臼。具有中等程度皮肤脆性增加、伸展过度和损伤倾向。
Ⅷ型(齿周围炎型):常染色体显性遗传,皮肤的Ⅲ型型胶原减少,特征为严重的牙周炎,牙周骨膜发育不良,30多岁时牙齿脱落,伴中度关节活动过度、皮肤脆性增加和轻度皮肤伸展过度。
Ⅸ型:即皮肤松弛-枕骨角综合征,为X连锁隐性遗传,其特征为皮肤松弛伸展、腹股沟疝、膀胱憩室、破裂、短臂、局限性旋前和旋后位、宽锁骨、枕骨角,为铜代谢异常伴赖氨酸氧化酶缺陷所致。
Ⅹ型:(纤维连接蛋白缺陷型)常染色体隐性遗传。临床表现是因为血小板凝集缺陷所致。当轻度碰伤时就可表现出显著的皮肤青紫。其他症状包括小关节显著活动过度、中等皮肤伸展过度。本型患者血浆中纤维连接蛋白存在功能上的缺陷。
组织病理
表皮角化过度,颗粒层增加,棘细胞层肥厚。用弹性纤维染色法可见真皮上部弹性纤维数量增加。真皮下部除数量增加外,还伴有肿胀、断裂等退行性改变。胶原纤维数量减少,纤维排列紊乱或成涡轮状。有时胶原纤维水肿。基质染色淡。真皮血管增多,血管壁增厚,血管四周有慢性炎细胞浸润。皮下脂肪组织减少。
诊断及鉴别
典型病例诊断一般不困难。但需与皮肤松弛症、Turner综合征和成骨不全进行区别。皮肤松弛症表现为皮肤松弛多皱,因此而产生皮肤悬吊,皮肤的弹性不增加,没有关节伸展过度。Turner综合征患者除皮肤弹性增加外,常伴有侏儒症、前额部斑状脱发、短颈、肘外翻、口呈三角形以及生殖器官发育不良等表现。成骨不全也是一种全身性结缔组织遗传病,累及骨骼、肌腱、韧带、筋膜等组织,两者往往合并发生。
治疗
本病无特效治疗方法,以预防外伤和对症治疗为主。症状明显的疝和憩室可作修补术。大血管破裂引起的急性出血性休克应作紧急手术处理。由于组织的脆性增加,常给缝合造成困难,缝合的伤口易裂开,因此,尽量避免不必要的手术。必要的手术缝合之后,应加压包扎,并延期拆线。维生素C能增强皮肤的抵抗力和促进伤口愈合,并增强肌肉强度,成人2~4g/d,儿童酌减。
