
本病又称红斑性皮肤松弛症(erythematous anetoderma),系由Jadassohn于1891年首先描述,Finger等称其为斑状萎缩性皮炎(dermatitis atrophicans maculosa)。
病因:尚不明了,有内分泌(性腺、甲状腺、脑下垂体)功能紊乱,神经营养及自主神经系统功能失调,外伤,感染,先天性家族因素及免疫因素等诸学说。孙永建报道一家族连续3代有1人患病。
临床症状
比较常见的为Jadassohn型,好发于青年女性,在20~30岁发病者约占一半,其特点为在皮肤发生萎缩之前局部先有炎症改变。开始直径为0.2~0.3cm,边界明显,圆形、椭圆形或不规则的铅红色至紫红色斑,逐渐增大,在1~2周内达到直径0.5~1.0cm或更大,此时皮损中央颜色开始变淡。经几周至几个月后,皮损表面逐渐变得光滑、干燥发亮、起皱,或为微凹的萎缩斑片,而边缘颜色不变,故形成环形,继续进展终于变成淡白或珍珠母色、柔软与松弛的扁平隆起呈软瘤状,如用指尖压之则下陷,抬起则复原,呈疝样表现。皮损好发于颜面、躯干,尤其是在上肢的伸侧或肩部,可单发或多发,多发性皮损分布常对称而呈播散性,损害之间皮肤正常,不累及黏膜。本病一般无自觉症状,但少数有瘙痒或灼热感。病程长,当皮损发展至一定程度后终生不变。
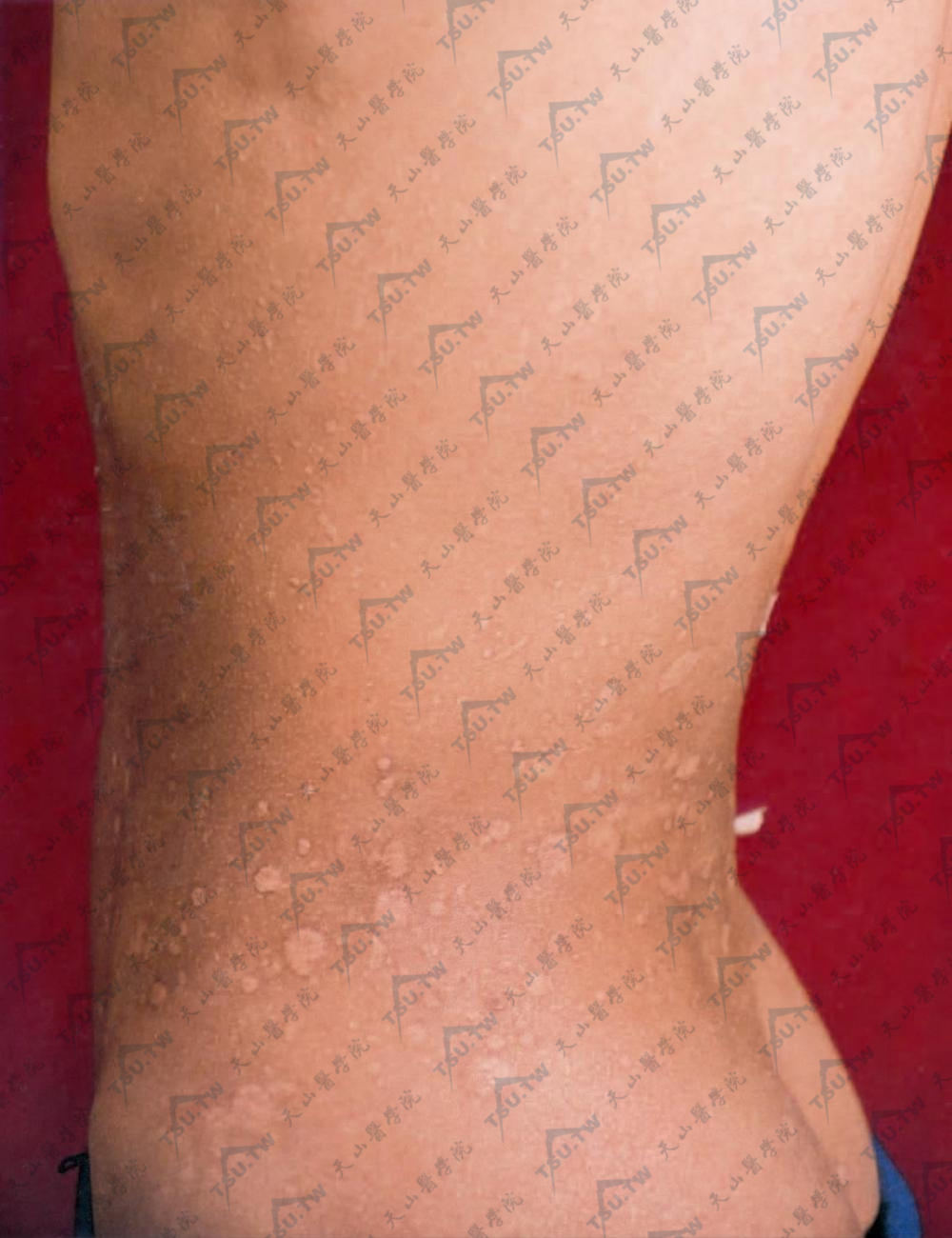
侧腹、腰背部有多个2~20mm圆形、卵圆形,光滑、干燥、微皱、微凹的萎缩斑片及柔软松弛的扁平隆起斑片,指尖接触时有嵌入疝孔的感觉
另有Pellizari型,罕见。先有风团样损害,经反复发作后形成柔软囊性疝样斑状萎缩。
组织病理
表皮萎缩,基底细胞层色素减少。真皮萎缩,胶原纤维变性,弹性纤维断裂、破坏或消失。在Jadassohn型中,真皮有炎症反应,在血管周围有淋巴细胞、中性粒细胞及嗜酸性粒细胞浸润。Oikarinen等用放射免疫法测定显示弹力蛋白有明显减少。
诊断及鉴别
根据卵圆形青白色斑、皮肤菲薄、光亮、起皱,指压可感觉其下有缺陷等,诊断一般不难。应与下列疾病相鉴别:
- 神经纤维瘤病幼年发病,皮疹不萎缩,常为隆起悬垂或形成大的赘瘤,伴有广泛的咖啡斑及中枢神经系统异常。
- 继发性斑状皮肤萎缩斑如梅毒、麻风、盘状红斑狼疮等,在发生皮肤萎缩斑前都应有典型的原发病史。
- 局灶性皮肤发育不全所致的脂肪疝。
- 白点病为较小的点状白色瘢痕性损害,做组织病理检查可鉴别。
皮损呈不规则集簇分布,起病年龄小,伴有其他先天性缺陷。
治疗
对Jadassohn型病人,在早期有炎症的阶段可试用青霉素治疗,但到萎缩期则治疗困难。已报道治疗本病的方法包括砷剂、维生素类、自血疗法、按摩、温浴、体疗、透热疗法、紫外线、糖皮质激素、甲状腺素及垂体激素等,但疗效均不能肯定。
